Integrated Bioinformatics Approach Unveils Genes Linked to Metastasis in Head and Neck Squamous Cell Carcinoma
|
Nalla Reddy Harsha Vardhan Department of Biotechnology, VFSTR deemed to be University, Guntur, India 522213. Department of Product and Process Development, NeoDx Biotech Labs Pvt Ltd, Bangalore, India 560099. Radhu Kantilal Ladani Department of Product and Process Development, NeoDx Biotech Labs Pvt Ltd, Bangalore, India 560099.
Aluru Ranganadha Reddy* Department of Biotechnology, VFSTR deemed to be University, Guntur, India 522213.
|
*E-mail: [email protected]
Abstract
The study discusses the challenges associated with the HNSCC metastasis and aims to identify hub genes related to the Cancer Stem Cells and metastasis in HNSCC through bioinformatics analysis. HNSCC is a prevalent type of cancer having a low five-year rate of survival, largely due to advanced stages at diagnosis and distant metastasis. The study hypothesizes that CTCs with CSC characteristics are responsible for metastasis. To investigate, we utilized mRNA-based stemness index (mRNAsi) and WGCNA on transcriptomic data by TCGA HNSCC cohort. The study involved multiple analytical steps, including DGE analysis, co-expression network, functional enrichment, hub gene identification, PPI network, and survival evaluation. The results revealed 18 hub genes associated with CSCs and metastasis in HNSCC. Further screening narrowed the hub gene list to nine genes, CDC45, MCM5, ASF1B, RFC4, E2F1, TK1, CHTF18, CENPM, and CDCA3.These genes have been discovered to be up-regulated in the samples of HNSCC compared to normal tissues. Highlighted for potential prognostic significance in HNSCC patients, CHTF18 and CDC45, with CDC45 playing a role in DNA replication, are associated with metastasis and CSC-like behavior. The study offers insights into potential hub genes associated with CSCs and metastasis in HNSCC. However, further experimental validation and clinical data integration are necessary to confirm these findings and establish clinical relevance. This study utilizes bioinformatics tools to identify genes influencing metastatic behavior in HNSCC, revealing potential therapeutic targets and underscoring the intricacies of cancer metastasis. Other hub genes are linked to various cancers, promoting invasion, proliferation, and migration.
Keywords: Head and neck squamous cell carcinoma, Metastasis, Hub genes, Cancer stem cells, mRNA-based stemness index, Weighted gene Co-expression network analysis
Introduction
Metastasis remains a major challenge in most solid tumors due to the lack of adequate treatment regimens and subsequent poor prognosis of the patients (Rus et al., 2020). HNSCC, the sixth most common cancer in the world, has a 50 % 5-year rate of survival, one of the major reasons being the advanced stage at presentation (Dahiya & Dhankhar, 2016). Patients who present having advanced disease are found to develop distant metastasis at a rate of twenty percent, which results in a poor prognosis for the patient (Schaeffer et al., 2014); the 5-year survival drops from 80% for the stage-I-II disease to about 20% in stage-III-IV. Distant metastasis rates in HNSCC range between 25-50%, the common sites being the brain, lungs, and bones, the rates of metastasis also differ between the various sub-sites of HNSCC (Nicoară et al., 2023; Radu et al., 2023). The current mode of detection of metastasis is imaging-based, PET scan be the best with a sensitivity of detecting 1-1.5 cm of tumor at the distant site, however, this does not provide for appropriate treatment management. Detection of patients with metastatic potential at presentation is a way forward to increase the rate of survival and patient’s treatment outcomes with advanced disease. Investigations down the decades have delineated the major processes underlying distant metastasis; dissemination of epithelial tumor cells into the blood, epithelial-mesenchymal transition (EMT), migration/invasion, and secondary tumor initiation. Circulating tumor cells (CTCs) in the bloodstream, considered to be the aggressive, migratory cells that disseminate from the primary tumor, are said to be the potential seeds that lead to distant metastasis. CTCs were majorly registered in the patient's peripheral blood with cancers of the breast, colorectal, lung, prostate, and oral cancer (Valiyaveedan et al., 2015; Kulsum et al., 2017) with associated prognostic efficacy, possibility of treatment surveillance, and disease progression (Maxime et al., 2016; Rutkowski et al., 2017; Lop et al., 2018; Matos et al., 2023). Cancer stem cells (CSCs) are known to attain EMT-like characteristics, with higher migratory and invasive capacity and retaining their tumorigenic property (Dahiya & Dhankhar, 2016). Further, reports suggesting the presence of non-migratory and migratory (Schaeffer et al., 2014) subgroups within CSC emphasize the need for a detailed investigation. Studies from the different groups have also shown that CSCs enriched by exposure to drugs do attain high migratory and invasive capacity, EMT marker profile, and increased properties of tumor initiation (Valiyaveedan et al., 2015; Kulsum et al., 2017). With the background of this evidence, we hypothesize that the CTCs with CSC characteristics are the ‘metastatic CTCs’ and the use of CSC-specific hub genes might provide insights into their accurate detection/targeting in HNSCC.
Over the past decade, the advancement of high-throughput technology has led to a substantial accumulation of data within public databases, furnishing a wealth of high-quality information for more in-depth exploration (Hajjo et al., 2021). The realm of oncological research has greatly benefited from the successful application of bioinformatics based machine-learning techniques. Among these, the One-Class Logistic Regression (OCLR) (Hemanth et al., 2020) stands out as a prominent method for encapsulating the prospects of stem cells. OCLR introduced the concept of a stemness index known as mRNAsi. In the context of this investigation, we delved into the clinical implications of mRNAsi and identified genes associated with CSCs (Cancer Stem Cells) or metastasis in HNSCC samples sourced from the TCGA dataset. This exploration was undertaken through the adept utilization of mRNAsi and WGCNA, harnessing the power of bioinformatics techniques. To execute this study, a variety of R-programming packages were employed, enabling a comprehensive analysis.
Materials and Methods
Software and R Packages
In this project, we utilized R version 4.3.1 running on the Windows platform. All the given R-packages were sourced from Bioconductor (Gentleman et al., 2004; Otasek et al., 2019), which provides a collection of open-source packages for bioinformatics research. The software and platform we employed are both open source and readily accessible.
Data Sources
The RNA-Seq data utilized in this research were sourced through TCGA, HNSC cohort, available at the UCSC database. The data set consisted of 500 primary site tumor samples, 2 metastatic samples, and 44 normal samples of tissue. Concurrently, relevant clinical data, including age, sex, life status, and stages, have been extracted through the same TCGA HNSC cohort. Subsequently, the expression values underwent log2 transformation (Ritchie et al., 2015). In instances where multiple expression values were available for a particular gene, we averaged the values to obtain are present active expression level. Additionally, the corresponding mRNAsi data, along with EREG-mRNAsi, were obtained from a referenced journal (Hemanth et al., 2020). To maintain data integrity, samples lacking clinical information and stemness index data were excluded from the subsequent analyses.
Correlation of the mRNAsi and Clinical Characteristics
The mRNAsi scores, based on the OCLR algorithm, were determined for stemless and stemness aspects, referred to as EREG-mRNAsi, and both scores ranged from 0 to 1. Our investigation involved a comparative analysis of mRNAsi across various aspects: normal vs. tumor group and tumor vs. metastasis group, conducted using an unpaired t-test. To assess significant changes in mRNAsi among different groups of variables, including treatment effects and different TNM (Tumor Node and Metastasis) stages, we utilized one-way ANOVA. All the above evaluations have been conducted by utilizing the GraphPad Prism version 8 (64-bit). For further analysis, the mRNAsi values (stemness index) were categorized into two subgroups as per the median cutoff value. We then conducted a Kaplan-Meier (K-M) evaluation for comparing the OS among the higher and lower subgroups, employing the log-rank test with the "survival" and "survminer" packages in R software. A p<0.05 was considered indicative of a statistically major difference.
Differential Gene Expression Analysis Statistically Significant Difference Was Presumed to Exist When the p< 0.05.
In this research, we employed the R edgeR (Robinson et al., 2010) package to conduct a comprehensive evaluation of DEGs (Mirza et al., 2019) among Tumor + Metastasis Vs Normal (T+M Vs N), Metastasis Vs Normal (M Vs N), Metastasis Vs Tumor (M Vs T). The identification of DEGs was carried out using the Wilcoxon signed-rank test. Our selection criteria for DEGs encompassed three key factors: a minimum absolute log2 fold change (FC) greater than 0.1, a p<0.05, and FDR (False Discovery Rate) below 0.05 for combinations M Vs N and T+M Vs N. For M vs. T a minimum absolute log2 fold change (FC) >F 0.1, a p-value less than 0.05, and an FDR below 0.34. To enhance the scope of our investigation and capture as many potential DEGs as possible between the sample groups, we adopted a criterion focused on low log fold change. Subsequently, we utilized the ensemble database R package "org.Hs.eg.db" to convert the Ensembl IDs of the differentially expressed genes into their corresponding gene names. To present our findings in a visually informative manner, we created volcano plots using the R-packages "ggplot" and "ggplot2”. These plots offered a clear representation of the DEGs identified in each of the examined groups.
WGCN Analysis
Co-Expression Network and Identification of Significant Modules
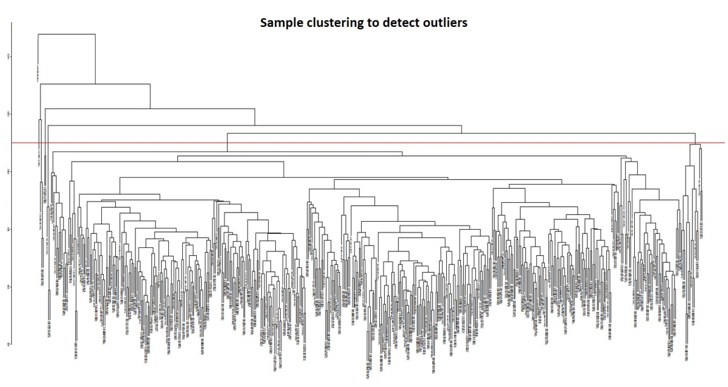
In this study, we constructed co-expression networks by utilizing the WGCNA package (Langfelder & Horvath, 2008) in the R-software (v4.3.1), based on the DEGs attained in the former step. The goodSamplesGenes function was employed to verify the completeness of the sample data and identify any outliers through sample clustering, which were subsequently removed. Pearson correlation coefficients were calculated between groups of genes and their absolute values have been utilized to create a gene expression similarity matrix by utilizing the mentioned formula: aij=|cor(xi,xj)|^β, xi&xj denotes the nodes i&j of the network, correspondingly. The parameter 'β' was carefully chosen to ensure the gene distribution adhered to a scale-free network based on connectivity. With the chosen 'β' value, we made the adjacency matrix and the TOM (Topological Overlap Matrix) (Pei et al., 2020). The TOM has been then subjected to gene dissimilarity-based clustering, and the resulting trees have been divided into distinct modules utilizing the dynamic shear approach. Each module was required to contain at least 50 genes. To enhance module identification, we combined some modules based on correlation coefficients. From the pool of modules, we have chosen the hierarchical clustering module which exhibited the closest connection to mRNAsi & EREG-mRNAsi for additional investigation. To gauge the importance of genes within each module, we utilized Module Eigengene (ME), Module Significance (MS), and Genetic Significance (GS). GS was determined by correlating gene expression with mRNAsi and EREG-mRNAsi, employing a log10 to transform the p-value in linear regression. This approach sheds light on the significance of every gene to specific traits. MS represented the average significance of all genes within a module, providing an overall measure of the module's importance. By merging similar modules by utilizing a cut-off value of 0.25, we identified modules having a great MS, deeming them most pertinent to sample traits. The first principal component discovered by evaluating the principal component on the gene expression matrix for every module is known as ME. The module with the greatest MS was chosen for further study because it was thought to be connected with mRNAsi and EREG-mRNAsi. Following the identification of major modules, we computed the GS and module membership (MM), which represents the association among the genes that make up a module also the gene expression patterns. To determine hub genes related to stemness indices, we set the inclusion criteria as follows: MM > 0.6 and correlation between gene GS > 0.4 and 0.1.
Functional Enrichment Analysis
Functional enrichment for GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) evaluates the module that is selected using the clusterProfiler package (Yu et al., 2012; Aschenbrenner et al., 2021). The GO evaluation involved three terms: molecular function (MF), cellular component (CC), and biological process (BP). To filter the results, the thresholds were set as follows: a modified P< 0.05 and a q<0.05.
Relationships and Interactions Among Hub Genes
The network of protein interactions may be studied and visualized using the online database STRING. Genes with a score of less than 0.4 in the STRING database were selected to create a network model that Cytoscape display (v3.10.0). CytoHubba, a plugin for Cytoscape, determined the MNC, MCC, Degree, and EPC of each node. The genes with the highest MNC, MCC, Degree, and EPC values in this analysis were regarded as hub genes.
Correlation of Hub Genes with Survival Data
The TCGA database's tumors and normal tissues analyzed for differential expression profiling, patient survival, and pathological staging using the web tool GEPIA2. To make up for the dearth of normal tissue samples in the TCGA database, GEPIA data are additionally taken from the sequencing data of normal tissues in the Genotype-Tissue Expression (GTEx) project 6 (Tang et al., 2017). To determine if the hub gene was differentially expressed in various TNM stages and whether its expression was greater in tumor tissue than in normal tissue, this database was employed. The hub genes were also analyzed using HPA (Human Protein Atlas).
Verification of the Hub Genes Using Human Protein ATLAS (HPA)
Immunohistochemistry (IHC) by the HPA database has been utilized for comparing the survival-related gene protein expression between HNSCC and normal tissues. For researchers, HPA is a useful database that offers a significant quantity of proteomics and transcriptomic data in certain human tissues and cells. Additionally, the most used method of using immune staining for determining the relative amount as well as the protein location is the IHC based on a pattern of protein expression.
Results and Discussion
mRNAsi Index is Significantly Associated with the HNSCC
We obtained the clinical data and transcriptome profiling for 546 Samples. The dataset included 44 samples of normal tissue, 2 samples from metastatic disease, and 500 samples of tumors. Age, gender, status of life, length of survival, illness stage, and classification of TNM (Tumor-Node-Metastasis) stage are all included in the clinical information. The mRNAsi index of each instance was obtained from the appendix of Malta's paper (Huang et al., 2019) and was then combined with the dataset from the TCGA. A total of six TCGA samples without mRNAsi information were excluded from the study.
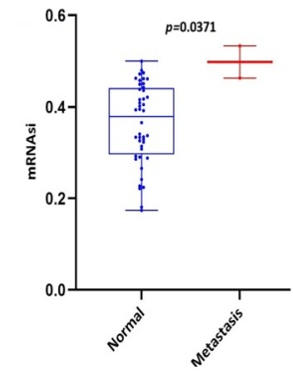
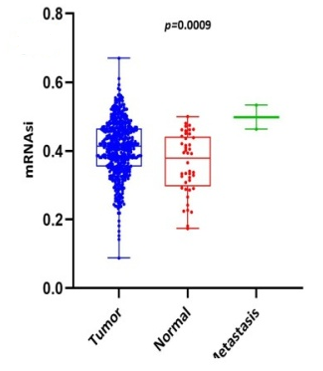
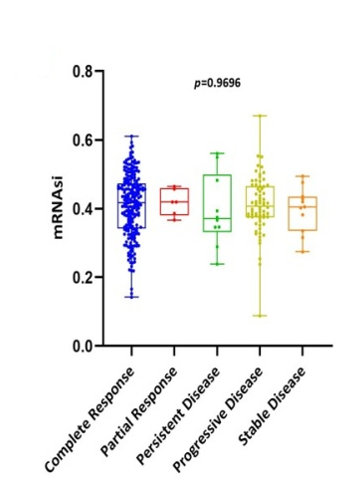
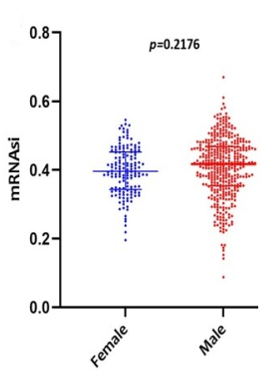
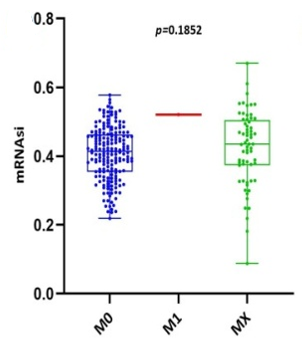
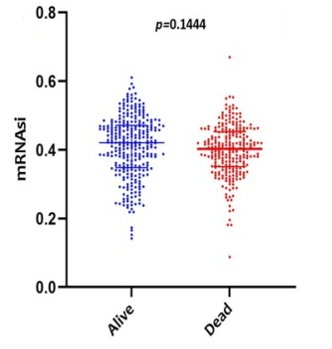
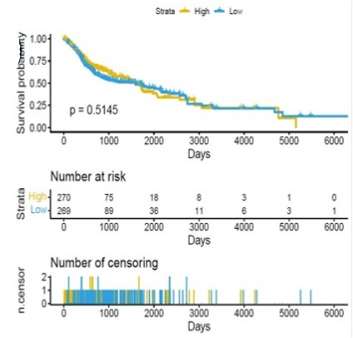
Scatter plots were created, as shown in Figure 1, to show how the global mRNAsi profiles and clinical characteristics correlate. The difference in the mRNAsi between normal and HNSCC tumors is 44 and 494 samples respectively (Figure 1a). The difference in the mRNAsi among metastasis is 2 samples and for normal it is 44 samples (Figure 1b). The difference in the mRNAsi between metastasis is 2 samples and the tumor is 494 samples (Figure 1c). A comparison of mRNAsi between Tumor samples (494), normal samples (44), and metastasis samples (2) (Figure 1d). Variations in the mRNAsi according to the treatment impact; complete responder 217 samples, Partial responder 6 samples, stable disease 11 samples, progressive disease 10 samples, and potentially resettable disease 68 samples (Figure 1e). Variations in the mRNAsi among the male 394 samples and Female146 samples (Figure 1f). Comparison of the mRNAsi among 4 various T-stages (T0, 1 sample; T1, 48 samples; T2, 150 samples; T3, 103 samples and T4, 183 samples; Tx, 33 samples (Figure 1g). By comparing the mRNAsi among the 4 various N-stages (N0, 185 samples; N1, 70 samples; N2, 173 samples; and N3, 7 samples; Nx, 80 samples) (Figure 1h). Comparison of the mRNAsi among various M stages (M0, 187; M1, 1; Mx, 64) (Figure 1i). Comparison of The mRNAsi among 4 various stages of Tumor, stage I 27 samples; stage II 85 samples; stage III 84 samples and stage IV 276 samples (Figure 1j). Comparison of the mRNAsi between the vital status (Alive 292 samples; Dead 248) (Figure 1k). Kaplan–Meier survival curves depicted the low mRNAsi group having a good rate of OS in comparison to the high mRNAsi group (Figure 1l).
|
|
|
a) |
|
|
|
b) |
|
|
|
c) |
|
|
|
d) |
|
|
|
e) |
|
|
|
f) |
|
|
|
g) |
|
|
|
h) |
|
|
|
i) |
|
|
|
j) |
|
|
|
k) |
|
|
|
l) |
|
Figure 1. Scatter plots |
Differential Gene Expression Analysis
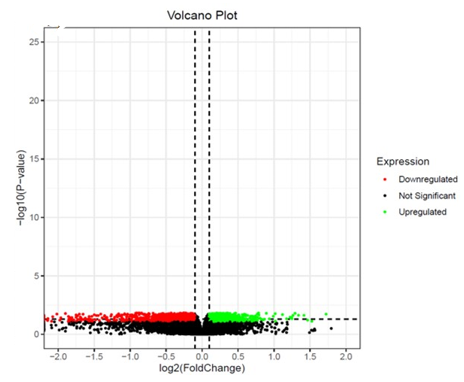
Differential genes observed with Tumor, metastasis Vs Normal 2532 genes (up-regulated 1244, down-regulated 1288) (Figure 2a). Differential genes observed between Metastasis Vs Normal 994 (up-regulated 618, down-regulated 376) (Figure 2b). Differential genes observed between Metastasis Vs Tumor 2823 (up-regulated 1397, down-regulated 1426) (Figure 2c).
|
|
|
a) |
|
|
|
b) |
|
|
|
c) |
|
Figure 2. Differential gene expression analysis |
WGCNA
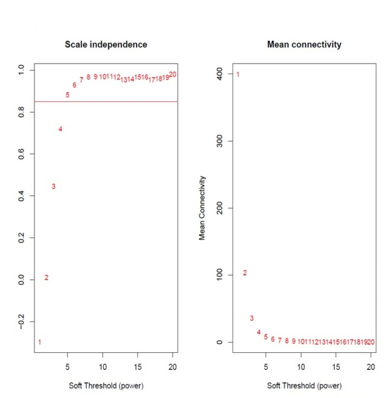
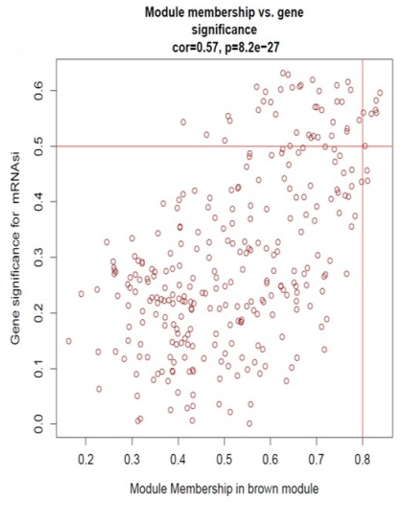
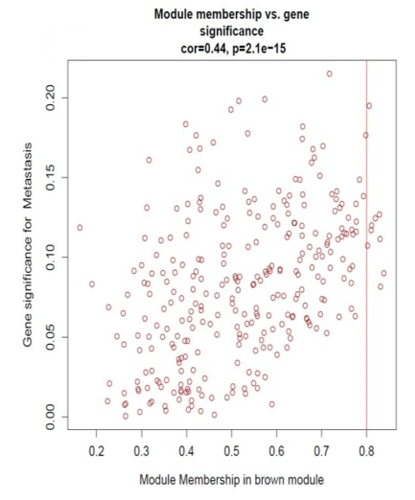
Identification of modules associated with clinical information from the TCGA HNSCC patient sample data. After eliminating 25 outlier samples, the co-expression network included all DEGs (Figure 3a). β=5 met the soft threshold parameter of the construction requirements for scale-free distribution, and the curve reached R2 = 0.85 (Figure 3b). To combine the comparable modules, the MEDiss Threshold was adjusted to 0.45, and 9 modules were eventually acquired (Figure 3c). The relationships between the modules and the characteristics of mRNAsi and metastasis were then assessed. By the mRNAsi and metastasis correlation coefficient R2, we selected the positively correlated mRNAsi Brown (R2=0.57; P-Value = 2e-41) module and metastasis (R2 =0.15; P-Value = 0.001) module (Figure 3d). Scatter plot of module eigengenes in the brown co-expression modules for mRNAsi and metastasis (Figures 3e and 3f).
|
|
|
a) |
|
|
|
b) |
|
|
|
c) |
|
|
|
d) |
|
|
|
e) |
|
|
|
f) |
|
Figure 3. Weighted Gene Co-Expression Network Analysis |
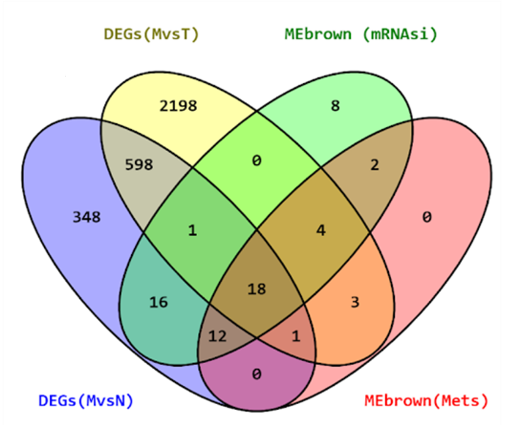
For identifying hub gens the threshold for mRNAsi used is (GS>0.4 and MM>0.6) and for metastasis (GS>0.1 and MM>0.6). To identify the overlapping hub genes between mRNAsi Brown module, Metastasis Brown module, DEGs of M vs. T, and DEGs of M vs N, we found 18 common hub genes (Figure 4a).
|
|
|
a) |
|
|
|
b) |
|
|
|
c) |
|
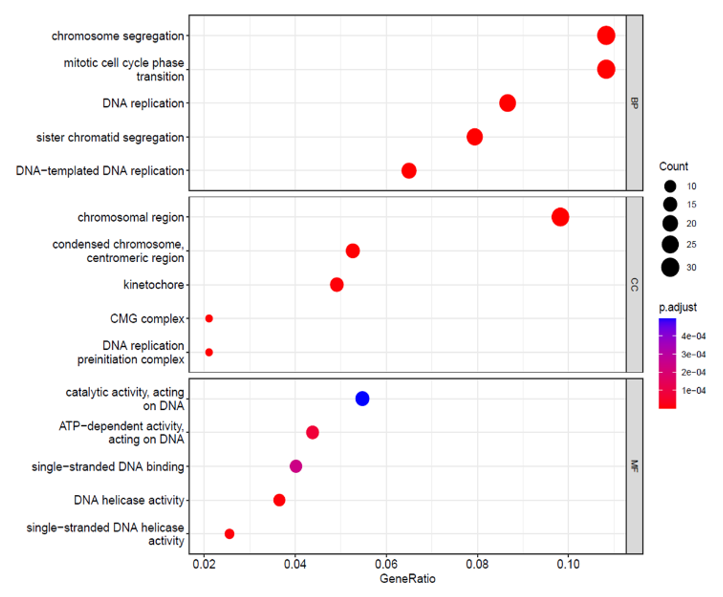
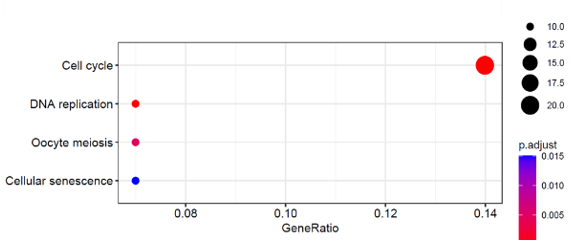
Figure 4. a) The Venn diagram of genes among two DEGs (M Vs T); (M Vs N) and co-expression modules MEbrown (mRNAsi and Mets). 4 b) c) Functional Enrichment Analysis. |
Functional Enrichment Analysis
GO and KEGG pathway enrichment studies have been conducted for the modules that have been closely associated with the metastasis and mRNAsi, and the top 5 enriched outcomes are shown in (Figure 4b). The brown modules, that show the strongest positive correlation with the metastasis and mRNAsi, are greatly enriched in nuclear division, DNA geometric change, DNA duplex unwinding, BP (Biological Process), chromosomal region, kinetochore, condensed chromosome centromeric region, CMG complex and DNA replication pre-initiation complex under cellular composition (CC) and ATP-dependent activity, DNA helicase activity, acting on DNA, single-standard DNA binding and DNA replication origin binding Molecular Functions (MF). According to the outcome of the KEGG evaluation (Figure 4c), the DEGs have been mostly concentrated in the replication of DNA, oocyte meiosis, cell cycle, and cellular senescence.
PPI Network Construction and Screening of the Hub Genes
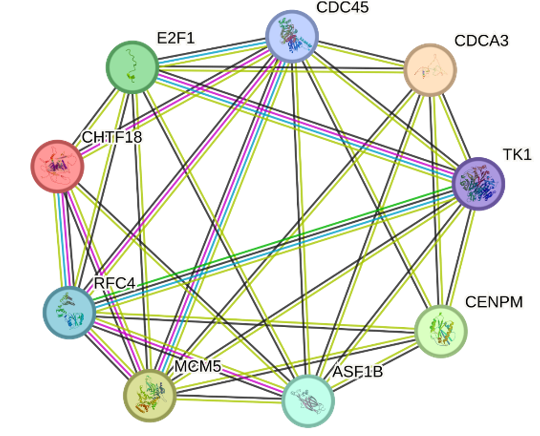
To further screen the hub genes, we have constructed a PPI network by utilizing the STRING (Search Tool for the Retrieval of Interacting Genes). For the common hub genes (Figure 4a) 18 nodes, and 59 edges have been created in the network, and the PPI improved the P-Value:<1.0e-16 (Figure 5a).
|
|
|
a) |
|
|
|
b) |
|
Figure 5. a) Protein interaction network of hub genes. b) Cytoscape (v3.10.0) was used to identify the top 10 genes using 4 algorithms and when comparing genes from these four algorithms 9 most common hub genes were revealed. |
To create a network module that Cytoscape could see, the genes with a score of less than 0.4 were selected (v3.10.0). A plug-in called CytoHubba is used to compute the nodes. We utilized all four algorithms: Degree, MNC, MCC, and EPC. when comparing the top 10 genes between these algorithms 9 genes were found to be common in all and were considered as the top hub genes CDC45, MCM5, ASF1B, RFC4, E2F1, TK1, CHTF18, CENPM, and CDCA3 (Figure 5b).
Survival Analysis
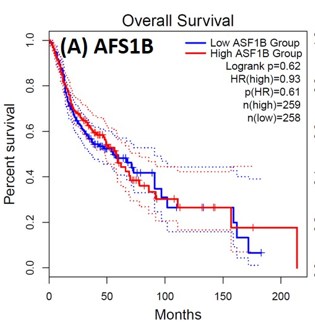
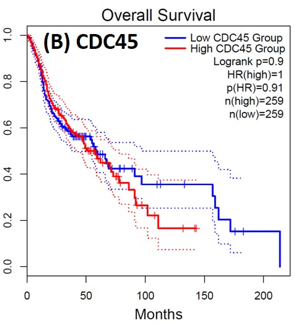
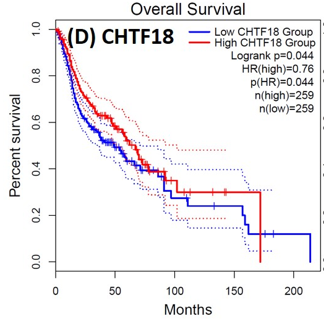
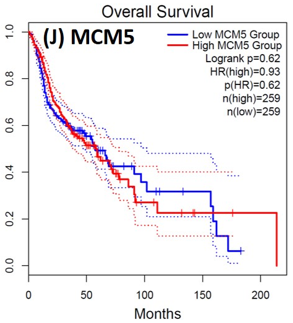
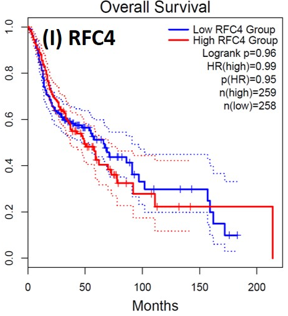
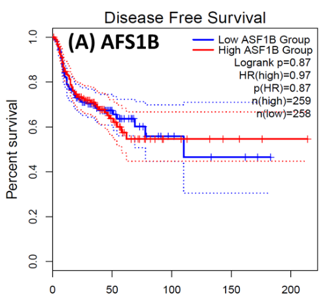
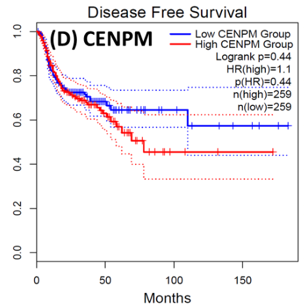
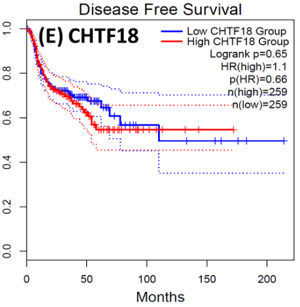
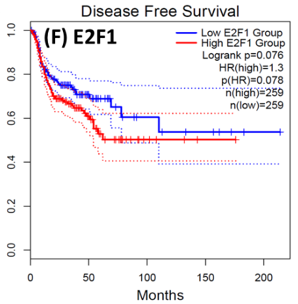
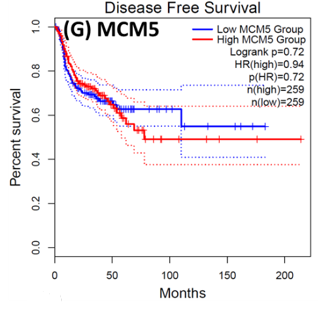
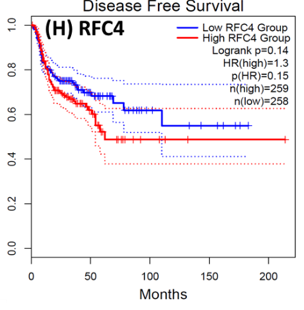
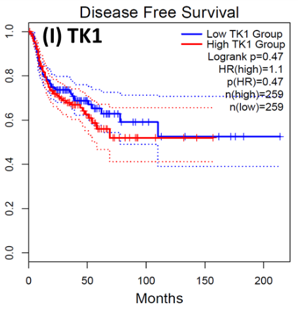
Following the CytoHubba plugin's screening of the nine hub genes (CDC45, MCM5, ASF1B, RFC4, E2F1, TK1, CHTF18, CENPM, and CDCA3). We checked the hub genes levels of expression in the TCGA database's patient database. When compared to normal tissues, all nine of the hub genes are highly up-regulated in HNSCC carcinoma. To examine the predictive significance of the genes in the patients of HNSCC, the Kaplan-Meier plotter has been utilized to conduct OS, DFS, and pathological stage evaluation of the 9 hub genes by utilizing the R-survival package as well as GEPIA2 database (Figure 6).
|
|
|
a) |
|
|
|
b) |
|
|
|
c) |
|
|
|
d) |
|
|
|
e) |
|
|
|
f) |
|
|
|
h) |
|
|
|
j) |
|
|
|
l) |
|
Figure 6. Kaplan-Meier plots and log-rank tests were employed on the GEPIEA2 server to assess the prognostic significance (OS) of hub genes within the TCGA HNSCC dataset. The dashed lines represent the upper and lower confidence intervals. |
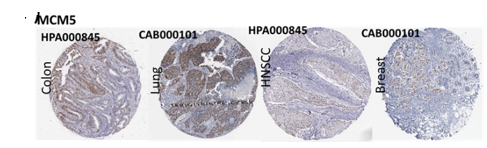
Of the 9 hub genes, the Kaplan–Meier evaluation proposed that the greater expression level of CHTF18 has been majorly linked with the poor OS of the patients of the HNSCC (P<0.044) (Figure 6d), and for CDC45 a significant difference was observed in the DFS (Figure 7b) in HNSCC patients. Furthermore, the protein expression levels of MCM5, RFC4, CDC45, ASF1B, E2F1, and TK1 (Figures 7 J,H) show moderate to high expression of HNSCC and other cancers.
|
|
|
a) |
|
|
|
b) |
|
|
|
c) |
|
|
|
d) |
|
|
|
e) |
|
|
|
f) |
|
|
|
g) |
|
|
|
h) |
|
|
|
l) |
|
|
|
J) |
|
|
|
H) |
|
Figure 7. Kaplan-Meier plots and log-rank tests were employed on the GEPIEA2 server to assess the prognostic significance (DFS) of hub genes within the TCGA HNSCC dataset. The dashed lines represent the upper and lower confidence intervals. (J,H) Immunohistochemistry images and expression levels in different cancers are shown for the hub genes using the Human Protein Atlas (HPA). |
HNSCC is a group of malignant tumors that arise in the mucosal linings of the oral cavity, pharynx, and larynx. Despite advancements in treatment modalities, HNSCC continues to pose a significant clinical challenge due to its high potential for metastasis. In recent years, research has focused on understanding the underlying metastasis mechanisms as well as the role of CSCs in driving this process. In this research, we utilized a bioinformatics method to identify the hub genes that have CSC sand involved metastasis using mRNA stemness index data. When performing differential analysis, we found 18 hub genes and when further evaluated with PPI and Cytohub we identified 9 hub genes (CDC45, MCM5, ASF1B, RFC4, E2F1, TK1, CHTF18, CENPM, and CDCA3) which are all up-regulated in HNSCC TCGA data. Among these nine hub genes, CHTF18 and CDC45 have shown high significance in OS and DFS in HNSCC patients. Where the expression of MCM5, RFC4, CDC45, ASF1B, E2F1, and TK1 in the HPA data base is moderate to high in Neck and head cancer and other cancers (Seo & Kang, 2018). The CDC45 gene, known for its role in DNA replication, is now implicated in cancer metastasis and stem cell-like behavior. Increased CDC45 expression in hepatocellular carcinoma (HCC) has been associated with larger tumor size and advanced stage, potentially facilitating metastasis by promoting epithelial-mesenchymal transition (EMT) (Yang et al., 2020; Yang et al., 2021). Additionally, CDC45 appears to influence cancer stem cell (CSC)-like behavior, as observed in gastric cancer cells where CDC45 .knockdown reduced the CSC-like population (Huang et al., 2019; Rommasi, 2023). CDC45 and MCM5 were reported for their interaction in the human replicative helicase, where CDC45 is termed as the guard for the MCM5 (Petojevic et al., 2015; Seo & Kang, 2018; Nowinska et al., 2019; Schmit & Bielinsky, 2021). ASF1B enhances migration, invasion, and cell proliferation by influencing the P53-mediated EMT signaling pathway in cancer of the lung, male thyroid cancer, renal cancer, and pancreatic adenocarcinoma. This highlights the potential of targeting ASF1B for effective cancer therapy (Jiangqiao et al., 2019; Qiu et al., 2022; Wang et al., 2022; Huang et al., 2023). RFC4 is a DNA replication factor in NSCLC it promotes metastasis and stemness properties when it couples with NICD1 (Liu et al., 2021). Transcriptomic analysis of tumors lacking the E2F1 gene revealed its significance as a central controller of several genes promoting metastasis in human breast cancer. Moreover, studies unveiled certain genes influenced by E2F1 that have uncharted functions in pulmonary metastasis (Hollern et al., 2019). EF1 also has been reported for its progression and metastasis in melanoma and prostate cancer (Davis et al., 2006; Alla et al., 2010; Putzer et al., 2010; Marquardt et al., 2018; Jamroze et al., 2021; Singh et al., 2023). TK1 appears to play a role in the intricate procedure for melanoma metastasis and breast cancer metastasis by facilitating its spread through the deep lymphatic system and contributing to its progression. This suggests that TK1 might be involved in promoting the movement of tumor cells (CTC) through the primary tumor site to distant lymph nodes and potentially other parts of the body. This mechanism could be a critical factor in the aggressive nature of melanoma and breast cancer and its ability to establish secondary growths in distant locations (Wu et al., 2013; Del Re et al., 2019; Qian et al., 2023). CHTF18, an evolutionarily conserved protein involved in DNA replication, assumes vital functions in both mammalian fertility and the process of meiosis (Mukerji et al., 2016). Increased expression of CENPM promotes the metastatic spread of tumors through the mTOR/p70S6K signaling pathway in the context cancer of pancreatic (Zheng et al., 2020). Cell division cycle-associated protein 3 (CDCA3) plays a crucial role in bladder cancer (BLCA) as MYBL2, a critical transcription factor, binds to its promoter, transactivates it, and promotes BLCA cell proliferation and migration. Elevated MYBL2 levels in BLCA tissues are associated with clinic pathological parameters and poor cancer-specific survival. This study establishes CDCA3 as a downstream target of MYBL2 and highlights their collaboration with FOXM1 to activate Wnt/β-catenin signaling, ultimately driving BLCA malignancy. CDCA3 is also involved in the promotion of tumorigenicity and metastasis in many cancers (Dou et al., 2020; Yu et al., 2020; Liu et al., 2022).
Conclusion
This study has successfully employed mRNAsi and WGCNA techniques to identify pivotal hub genes within HNSCC, offering the potential to predict metastatic tendencies. These hub genes are acknowledged for their significance in diverse cancer prognoses and metastasis scenarios. However, it's important to acknowledge that this investigation primarily constitutes a bioinformatics analysis, and the absence of clinical trials is notable. Future research endeavors will be directed towards the accumulation of an expanded clinical dataset, Accompanied by the integration of pertinent clinical insights. This strategic approach aims to bolster the credibility of the hub gene’s association with HNSCC metastasis and CSC behavior. This will be achieved through a series of targeted cellular and molecular experiments, ultimately a factor in a more comprehensive understanding of the complex dynamics underlying HNSCC metastasis.
Acknowledgments: None
Conflict of interest: None
Financial support: None
Ethics statement: None
Alla, V., Engelmann, D., Niemetz, A., Pahnke, J., Schmidt, A., Kunz, M., Stephan, E., Marc, S., Dirk, K., & Putzer, B.M. (2010). E2F1 in melanoma progression and metastasis. Journal of the National Cancer Institute, 102(2), 127-133. doi:10.1093/jnci/djp458
Aschenbrenner, A.C., Mouktaroudi, M., Kraemer, B., Oestreich, M., Antonakos, N., Nuesch-Germano, M., Gkizeli, K., Bonaguro, L., Reusch, N., Baßler, K., et al. (2021). Disease severity-specific neutrophil signatures in blood transcriptomes stratify COVID-19 patients. Genome Medicine, 13(1), 1-25. doi:10.1186/s13073-020-00823-5
Dahiya, K., & Dhankhar, R. (2016). Updated overview of current biomarkers in head and neck carcinoma. World Journal of Methodology, 6(1), 77-86. doi:10.5662/wjm.v6.i1.77
Davis, J. N., Wojno, K. J., Daignault, S., Hofer, M. D., Kuefer, R., Rubin, M. A., & Day, M. L. (2006). Elevated E2F1 inhibits transcription of the androgen receptor in metastatic hormone-resistant prostate cancer. Cancer Research, 66(24), 11897-11906. doi:10.1158/0008-5472.CAN-06-2497
Del Re, M., Bertolini, I., Crucitta, S., Fontanelli, L., Rofi, E., De Angelis, C., Diodati, L., Cavallero, D., Gianfilippo, G., Salvadori, B., et al. (2019). Overexpression of TK1 and CDK9 in plasma-derived exosomes is associated with clinical resistance to CDK4/6 inhibitors in metastatic breast cancer patients. Breast Cancer Research and Treatment, 178(1), 57-62. doi:10.1007/s10549-019-05365-y
Dou, D., Ren, X., Han, M., Xu, X., Ge, X., Gu, Y., Wang, X., & Zhao, S. (2020). CircUBE2D2 (hsa_circ_0005728) promotes cell proliferation, metastasis and chemoresistance in triple-negative breast cancer by regulating miR-512-3p/CDCA3 axis. Cancer Cell International, 20, 454.
Gentleman, R. C., Carey, V. J., Bates, D. M., Bolstad, B., Dettling, M., Dudoit, S., Ellis, B., Gautier, L., Ge, Y., Gentry, J., et al. (2004). Bioconductor: Open software development for computational biology and bioinformatics. Genome Biology, 5(10), R80.
Hajjo, R., Sabbah, D. A., Bardaweel, S. K., & Tropsha, A. (2021). Identification of tumor-specific MRI biomarkers using machine learning (ML). Diagnostics, 11(5), 742. doi:10.3390/diagnostics11050742
Hemanth, D. J., Deperlioglu, O., & Kose, U. (2020). An enhanced diabetic retinopathy detection and classification approach using deep convolutional neural network. Neural Computing & Applications, 32(7398), 707-721. doi:10.1007/s00521-018-03974-0
Hollern, D. P., Swiatnicki, M. R., Rennhack, J. P., Misek, S. A., Matson, B. C., McAuliffe, A., Gallo, K. A., Caron, K. M., & Eran, R. A. (2019). E2F1 drives breast cancer metastasis by regulating the target gene FGF13 and altering cell migration. Scientific Reports, 9(1), 10718. doi:10.1038/s41598-019-47218-0
Huang, J., Li, Y., Lu, Z., Che, Y., Sun, S., Mao, S., Lei, Y., Zang, R., Li, N., Zheng, S., et al. (2019). Analysis of functional hub genes identifies CDC45 as an oncogene in non-small cell lung cancer - A short report. Cellular Oncology (Dordrecht), 42(4), 571-578. doi:10.1007/s13402-019-00438-y
Huang, W., Lin, T., Huang, L., Wu, J., Hong, J., Qiu, F., Tian, Y., & Wang, Y. (2023). miR-24-3p regulates epithelial-mesenchymal transition and the malignant phenotype of pancreatic adenocarcinoma by regulating ASF1B expression. Biochemical Genetics, 61(2), 742-761. doi:10.1007/s10528-022-10278-5
Jamroze, A., Chatta, G., & Tang, D. G. (2021). Androgen receptor (AR) heterogeneity in prostate cancer and therapy resistance. Cancer Letters, 518, 1-9. doi:10.1016/j.canlet.2021.06.006
Jiangqiao, Z., Tao, Q., Zhongbao, C., Xiaoxiong, M., Long, Z., Jilin, Z., & Wang, T. (2019). Anti-silencing function 1B histone chaperone promotes cell proliferation and migration via activation of the AKT pathway in clear cell renal cell carcinoma. Biochemical and Biophysical Research Communications, 511(1), 165-172. doi:10.1016/j.bbrc.2019.02.060
Kulsum, S., Sudheendra, H. V., Pandian, R., Ravindra, D. R., Siddappa, G. R. N., Chevour, P., Ramachandran, B., Sagar, M., Jayaprakash, A., Mehta, A., et al. (2017). Cancer stem cell mediated acquired chemoresistance in head and neck cancer can be abrogated by aldehyde dehydrogenase 1 A1 inhibition. Molecular Carcinogenesis, 56(2), 694-711.
Langfelder, P., & Horvath, S. (2008). WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics, 9, 559. doi:10.1186/1471-2105-9-559
Liu, L., Tao, T., Liu, S., Yang, X., Chen, X., Liang, J., Hong, R., Wang, W., Yang, Y., Li, X., et al. (2021). An RFC4/Notch1 signaling feedback loop promotes NSCLC metastasis and stemness. Nature Communications, 12(1), 2693. doi:10.1038/s41467-021-22971-x
Liu, W., Shen, D., Ju, L., Zhang, R., Du, W., Jin, W., Xiong, K., Wang, G., Qian, K., Zhang, Y., et al. (2022). MYBL2 promotes proliferation and metastasis of bladder cancer through transactivation of CDCA3. Oncogene, 41(41), 4606-4617. doi:10.1038/s41388-022-02456-x
Lop, J., Rigó, A., Codina, A., de Juan, J., Quer, M., & León, X. (2018). Prognostic significance of extranodal extension in head and neck squamous cell carcinoma cN0 patients with occult metastatic neck nodes. Significado pronóstico de la extensión extranodal en pacientes con carcinomas escamosos de cabeza y cuello cN0 con metástasis ganglionares ocultas. Acta Otorrinolaringologica Espanola, 69(3), 156-164. doi:10.1016/j.otoeng.2017.07.002
Marquardt, S., Solanki, M., Spitschak, A., Vera, J., & Pützer, B. M. (2018). Emerging functional markers for cancer stem cell-based therapies: Understanding signaling networks for targeting metastasis. Seminars in Cancer Biology, 53(Suppl. 1), 90-109. doi:10.1016/j.semcancer.2018.06.006
Matos, L. L., Guimarães, Y. L. M., Leite, A. K., & Cernea, C. R. (2023). Management of stage III oral cavity squamous cell carcinoma in light of the new staging system: A critical review. Current Oncology Reports, 25(2), 107-113. doi:10.1007/s11912-022-01353-6
Maxime, M., Genrich, T., Christian, S., & Yan, M. (2016). Extracapsular spread in head and neck squamous cell carcinoma: A systematic review and meta-analysis. Oral Oncology, 62, 60-71. doi:10.1016/j.oraloncology.2016.10.003.
Mirza, B., Wang, W., Wang, J., Choi, H., Chung, N. C., & Ping, P. (2019). Machine learning and integrative analysis of biomedical big data. Genes, 10(2), 87. doi:10.3390/genes10020087
Mukerji, B., Harris, A., Dia, F., Singh, T., & Berkowitz, K. (2016). Special research presentation: A role for CHTF18 in female fertility and ovarian aging, fertility and sterility. Science Direct, 106(3), e67. doi:10.1016/j.fertnstert.2016.07.198
Nicoară, N. D., Marian, P., Petriș, A. O., Delcea, C., & Manole, F. (2023). A review of the role of cognitive-behavioral therapy on anxiety disorders of children and adolescents. Pharmacophore, 14(4), 35-39.
Nowinska, K., Ciesielska, U., Piotrowska, A., Jablonska, K., Partynska, A., Paprocka, M., Zatonski, T., Marzena, P., & Piotr, D. (2019). MCM5 expression is associated with the grade of malignancy and Ki-67 antigen in LSCC. Anticancer Research, 39(5), 2325-2335. doi:10.21873/anticanres.13349
Otasek, D., Morris, J. H., Bouças, J., Pico, A. R., & Demchak, B. (2019). Cytoscape automation: Empowering workflow-based network analysis. Genome Biology, 20(1), 185. doi:10.1186/s13059-019-1758-4
Pei, J., Wang, Y., & Li, Y. (2020). Identification of key genes controlling breast cancer stem cell characteristics via stemness indices analysis. Journal of Translational Medicine, 18(1), 74. doi:10.1186/s12967-020-02260-9
Petojevic, T., Pesavento, J. J., Costa, A., Liang, J., Wang, Z., Berger, J. M., & Botchan, M. R. (2015). Cdc45 (cell division cycleprotein45) guards the gate of the Eukaryote Replisome helicase stabilizing leading strand engagement. Proceedings of The National Academy of Sciences USA, 112(3), 249-258. doi:10.1073/pnas.1422003112
Putzer, B. M., Steder, M., & Alla, V. (2010). Predicting and preventing melanoma invasiveness: advances in clarifying E2F1 function. Expert Review of Anticancer Therapy, 10(11), 1707-1720. doi:10.1586/era.10.153
Qian, J., Liao, J., Liu, Z., Chi, Y., Fang, Y., Zheng, Y., Shao, X., Liu, B., Cui, Y., Guo, W., et al. (2023). Reconstruction of the cell pseudo-space from single-cell RNA sequencing data with scSpace. Nature Communications, 14(1), 2484. doi:10.1038/s41467-023-38121-4
Qiu, W., Wu, X., Shi, H., Liu, B., Li, L., Wu, W., & Lin, J. (2022). ASF1B: A possible prognostic marker, therapeutic target, and predictor of immunotherapy in male thyroid carcinoma. Frontiers in Oncology, 12, 678025. doi:10.3389/fonc.2022.678025
Radu, C. C., Delcea, C., Plesa, A., & Rad, D. (2023). Transforming perceptions of drug consumption among youth through a cognitive-social-medico-legal educational approach. Pharmacophore, 14(4), 50-56.
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., & Smyth, G. K. (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research, 43(7), e47. doi:10.1093/nar/gkv007
Robinson, M. D., McCarthy, D. J., & Smyth, G. K. (2010). edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26(1), 139-140. doi:10.1093/bioinformatics/btp616
Rommasi, F. (2023). Identification, characterization, and prognosis investigation of pivotal genes shared in different stages of breast cancer. Scientific Reports, 13(1), 8447. doi:10.1038/s41598-023-35318-x
Rus, M., Matei, R., Sandu, M. L., Delcea, C., & Siserman, C. (2020). Emotional distress and coping strategies of health care workers during COVID-19 pandemic. Romanian Journal of Legal Medicine, 28(4), 442-450.
Rutkowski, R., Schuster, M., Unger, J., Seebauer, C., Metelmann, H. R., Woedtke, T. V., Weltmann, K. D., & Daeschlein, G. (2017). Hyperspectral imaging for in vivo monitoring of cold atmospheric plasma effects on microcirculation in treatment of head and neck cancer and wound healing. Clinical Plasma Medicine, 7(8), 52-57. doi:10.1016/j.cpme.2017.09.002
Schaeffer, D., Somarelli, J. A., Hanna, G., Palmer, G. M., & Garcia-Blanco, M. A. (2014). Cellular migration and invasion uncoupled: increased migration is not an inexorable consequence of epithelial-to-mesenchymal transition. Molecular and Cellular Biology, 34(18), 3486-3499. doi:10.1128/MCB.00694-14
Schmit, M., & Bielinsky, A. K. (2021). Congenital diseases of DNA replication: Clinical phenotypes and molecular mechanisms. International Journal of Molecular Sciences, 22(2), 911. doi:10.3390/ijms22020911
Seo, Y. S., & Kang, Y. H. (2018). The human replicative helicase, the CMG complex, as a target for anti-cancer therapy. Frontiers in Molecular Biosciences, 5, 26. doi:10.3389/fmolb.2018.00026
Singh, N., Khan, F. M., Bala, L., Vera, J., Wolkenhauer, O., Pützer, B., Logotheti, S., & Gupta, S. K. (2023). Logic-based modeling and drug repurposing for the prediction of novel therapeutic targets and combination regimens against E2F1-driven melanoma progression. BMC Chemistry, 17(1), 161. doi:10.1186/s13065-023-01082-2
Tang, Z., Li, C., Kang, B., Gao, G., Li, C., & Zhang, Z. (2017). GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Research, 45(W1), W98–W102. doi:10.1093/nar/gkx247
Valiyaveedan, S. G., Ramachandran, B., Iliaraja, J., Ravindra, D. R., James, B. L., & Kulsum, S. (2015). Acquisition of cancer stem cell behavior plays a role in drug resistance to combination chemotherapy and prognosis in head and neck cancer. Journal of Stem Cell Research & Therapy, 05(01), 261. doi:10.4172/2157-7633.1000261
Wang, W., Xiao, L., Pan, D., & Hu, L. (2022). ASF1B enhances migration and invasion of lung cancers cell via regulating the P53-mediated epithelial-mesenchymal transformation (EMT) signaling pathway. Neoplasma, 69(2), 361-369.
Wu, B. J., Li, W. P., Qian, C., Ding, W., Zhou, Z. W., & Jiang, H. (2013). Increased serum levels of thymidine kinase 1 correlate with metastatic sites in patients with malignant melanoma. Tumor Biology, 34(2), 643-648. doi:10.1007/s13277-012-0591-0
Yang, C., Xie, S., Wu, Y., Ru, G., He, X., Pan, H. Y., Wang, S., & Tong, X. (2021). Prognostic implications of cell division cycle protein 45 expression in hepatocellular carcinoma. PeerJ, 9, e10824. doi:10.7717/peerj.10824
Yang, S., Ren, X., Liang, Y., Yan, Y., Zhou, Y., Hu, J., Wang, Z., Song, F., Wang, F., Liao, W., et al. (2020). KNK437 restricts the growth and metastasis of colorectal cancer via targeting DNAJA1/CDC45 axis. Oncogene, 39(2), 249-261. doi:10.1038/s41388-019-0978-0
Yu, G., Wang, L. G., Han, Y., & He, Q. Y. (2012). ClusterProfiler: an R package for comparing biological themes among gene clusters. Omics: A Journal of Integrative Biology, 16(5), 284-287. doi:10.1089/omi.2011.0118
Yu, J., Hua, R., Zhang, Y., Tao, R., Wang, Q., & Ni, Q. (2020). DNA hypomethylation promotes invasion and metastasis of gastric cancer cells by regulating the binding of SP1 to the CDCA3 promoter. Journal of Cellular Biochemistry, 121(1), 142-151. doi:10.1002/jcb.28993
Zheng, C., Zhang, T., Li, D., Huang, C., Tang, H., Ni, X. F., & Chen, B. (2020). Upregulation of CENPM facilitates tumor metastasis via the mTOR/p70S6K signaling pathway in pancreatic cancer. Oncology Reports, 44(3), 1003-1012. doi:10.3892/or.2020.7673