Structure-based Multi-targeted Molecular Docking and Molecular Dynamic Simulation Analysis to Identify Potential Inhibitors against Ovarian Cancer
Bandar Hamad Aloufi*
Abstract
Ovarian Cancer (OC) is among the most prevalent cancers in females. OC is one of the deadliest and worst prognosis diseases. Currently, there are no approved OC screening tests or early detection methods. Hence, new screening, prevention, and early detection strategies are still highly demanded. Plant compounds have recently become increasingly important in developing new, effective, and affordable anti-cancer drugs. To investigate the protein-ligand interaction, molecular docking was used with the Molecular Operating Environment (MOE) tool to find the best inhibitor for the target proteins. Compound spatial affinity for the active sites of the NY-ESO-1, RUNX3, and UBE2Q1 proteins was calculated using molecular docking. ADMET analysis was used to determine the drug-likeness of the selected compounds, while MD simulation and MMGBSA/MMPBSA experiments were used to further understand the binding behaviors. Pre-clinical tests can help confirm the validity of our in silico studies and determine whether the compound can be used as an anti-cancer drug to treat OC.
Keywords: Ovarian cancer, Anti-cancer drugs, NY-ESO-1, RUNX3, UBE2Q1
Introduction
OC is the deadliest gynaecological cancer and the fifth most common cause of cancer death among females. During her lifetime, 1 out of every 54 women will develop OC (Ghilardi et al., 2022). OC is classified into three types based on the origin of the cells: epithelial, stromal, and germ cell. Despite advances in therapeutic and diagnostic procedures, OC has the lowest survival rate of any gynaecological malignancy in developed nations (Siegel et al., 2016; Torre et al., 2018; Manchanda, 2022). Furthermore, it is critical to investigate the role of the tumor-causing microenvironment during the proliferation, early stages, and metastasis. Hence, it is critical to comprehend the root cause from various perspectives, such as molecular pathogenesis, histological subtypes, hereditary factors, epidemiology, treatment methods, and diagnostic perspectives (Auner et al., 2010; Ledermann et al., 2013; Kbirou et al., 2022).
OC has a low survival rate and poor prognosis due to a lack of early screening methods and ineffective treatments for advanced stages of the disease. The high mortality rate from OC is because 30% of advanced-stage tumors do not respond to standard chemotherapy, and most responders relapse over time (Auner et al., 2010; AlHussain et al., 2022; Haque et al., 2022). Patients with OC typically experience recurrence and progression of the disease due to resistance to current chemotherapy treatment. Most chemotherapy drugs prescribed are synthetically derived and toxic to cancer cells and normal cells (Gupta et al., 2001; Alhazmi et al., 2022; Pahwa et al., 2022). On the other hand, recent findings show that naturally extracted phytochemicals from plants have significant selective cytotoxicity for cancer cells while having minimal toxicity for normal cells. This may be a viable cancer treatment option (Devi et al., 2015; Roy et al., 2022).
Therefore, in our study, 2500 natural compounds of plant origin were collected and selected for their inhibitory properties. The compounds screened for docking were first put via in silico docking and high-throughput virtual screening before being chosen as the best inhibitor for the target proteins (NY-ESO-1, RUNX3, and UBE2Q1). The study of their interactions with the target proteins may assist in developing novel drugs for OC treatment.
Materials and Methods
Retrieval and Refinement of OC Proteins
Crystallized structures of NY-ESO-1, RUNX3, and UBE2Q1 were retrieved from Protein Data Bank (PDB) (Bittrich et al., 2022). Molecular Operating Environment (MOE) was used to prepare the retrieved structures for docking (Vilar et al., 2008; Wang et al., 2021).
Ligand Database Preparation
An extensive literature search was carried out to find phytochemicals that have been reported to be effective against OC. Phytochemical chemical structures were obtained from the MPD3 database, the Pubchem database, the MAPS database, and the Zinc database (Irwin & Shoichet, 2005; Kim et al., 2016; Mumtaz et al., 2017; AlShehri et al., 2022) in various ligand file formats, including mol, sdf, and mol2. These ligand structures were optimized with the Protonate3D module by adding partial charges in MOE. The MMFF94X force field was utilized to minimize each ligand’s energy. The ligands were then individually added to the MOE ligand database for docking purposes.
Binding Site Analysis
Active sites of the targeted receptors were predicted through the CPORT tool present in the Haddock interphase (de Vries & Bonvin, 2011; Remizova et al., 2022). These active site pockets are where the ligand will bind to the target receptors to inhibit its activity. The sites consist of hydrophilic, hydrophobic, donor, acceptor and metal-binding domains. The best predicted binding pocket was chosen for performing molecular docking.
Molecular Docking
Within the specified docking sites of the NY-ESO-1, RUNX3, and UBE2Q1 proteins, the MOE Dock tool was utilized to dock a ligand database of 2500 phytochemicals. The default ligand placement method was used to find 1000 best poses of docked molecules by the triangular matcher algorithm (Podvinec et al., 2010; Alhumaydhi et al., 2021; Burlaka, 2021; Kumar et al., 2022). The London dG scoring function was used to rescore simulated poses. The Force field refinement algorithm that applies the Generalized Born solvation model to calculate final binding energy while keeping receptor residues rigid was used to further minimize the top ten ranked poses per molecule created by London dG (Li et al., 2022). Binding affinity, S-score, and Root-Mean-Square Deviation (RMSD) values were used to rank all compounds. The goal was to select only those compounds for further investigation from the top-ranked poses that bind to active OC protein residues with a good dock score.
Analysis of Ligand- Receptor Interaction
To clearly understand the best-docked complex receptor-ligand interactions, MOE's LigX tool was utilized for analyzing ligand-receptor interactions in 2D plots (Khalifa et al., 2020; Alhumaydhi et al., 2021). It creates electrostatic, hydrophobic, Van der Waals forces in the active site of OC proteins and a two-dimensional graph of hydrogen bonding, contributing to drug-like molecule affinity. Using UCSF Chimera, the 3D images of OC protein inhibitor complexes were created (Pettersen et al., 2004; Pettersen et al., 2021).
Drug scan/ADME Toxicity
A computational approximation of the drug probability of docked phytochemicals was found through the drug scanning tool on the Molinspiration server within limits set by Lipinski's Rule. Using the ADMETsar server, the deposition, absorption, excretion, metabolism, and toxicity profiles of these hits were virtually evaluated (Cheng et al., 2012). Carcinogenic potential and AMES toxicity of inhibitors were also evaluated.
Free Energy Calculation Binding
The free energies binding of receptor-ligand complexes was calculated by the Schrodinger suite's Prime module and the OPLS-2005 force field (Mishra & Singh, 2022). The free energy binding was calculated by subtracting the complex free energy from the ligand and protein free energies sum (Ikot et al., 2020).
|
ΔG(binding) = ΔG(complex) – [ΔG(ligand)+ ΔG(protein)] |
(1) |
where ΔG(binding) denotes the binding free energy and ΔG(complex), ΔG(ligand), and ΔG(protein) denote the free energies of the complex, ligand, and protein, respectively. Using the MM-GBSA method and the MM-PBSA.py program, with 250 snapshots taken at equal intervals over the last 20 ns of simulation, binding free energies were estimated.
MD Simulation
The Desmond software was used to run MD simulations (Release, 2017). To determine protein-ligand interactions, which were solvated using the simple point charged (TIP4P) water model, the optimized potentials for liquid simulations (OPLS)-2005 force field was used in this system. A 10Å buffer region was created among the box sides and protein atoms using the orthorhombic water box. Na+ ions neutralized the systems after overlapping water molecules were removed. The energy was calculated using the OPLS-2005 force field. The temperature was kept constant at 300 K during the integration step, yielding a 2.0 fs value. Finally, for monitoring the consistency of the OC proteins in their native motion, the Root Means Square Deviation (RMSD), Root Mean Square Fluctuations (RMSF), Solvent Accessible Surface Area (SASA), and Radius of Gyration (RoG) were calculated. For up to 100 ns, the synchronize file was saved every 5000 ps, and the result was examined using Nagasundaram et al.'s method (Heimes et al., 2022; Kabir et al., 2022).
Results and Discussion
Proteins Preparation
The study began with retrieving the crystal structures of NY-ESO-1, RUNX3, and UBE2Q1 proteins from the PDB database with PDB IDs of 1S9W, 5W69, and 2QGX, respectively. X-ray diffraction was used to determine the structure up to a resolution of 2.20 Å, 2.80 Å, and 2.56 Å, respectively. MOE was used to prepare the structures for docking. 3D protonation, removal of water molecules, and energy minimization were performed using MOE.
Binding Site Analysis
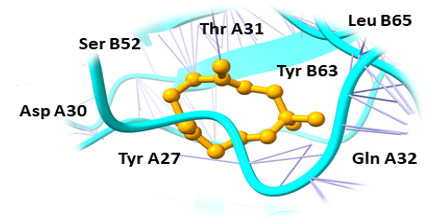
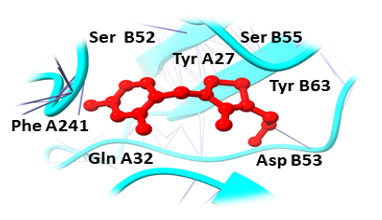
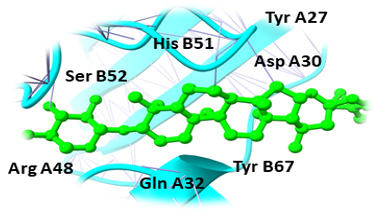
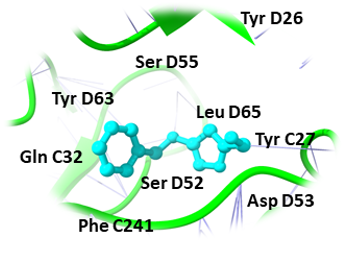
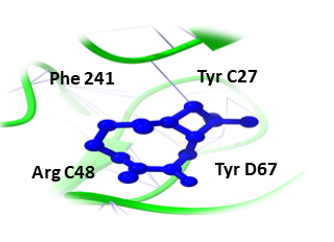
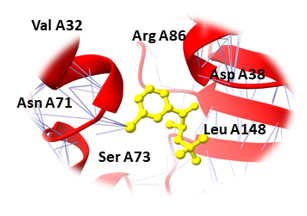
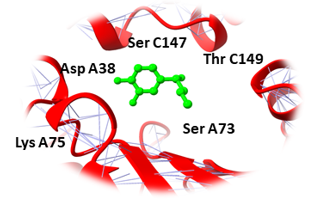
Binding pockets against the target receptors were chosen from Cport. For receptor NY-ESO-1, the reported active residues were Tyr A27, Asp A30, Thr A31, Gln A32, Arg A48, Ser B52, Asp B53, Ser B55, Tyr B63, Leu B65, Tyr B67, Phe A241. Similarly, for RUNX3, it was Tyr D26, Tyr C27, Gln C32, Ser D52, Asp D53, Ser D55, Tyr D63, Leu D65 and Phe C241. While Val A32, Asp A38, Asn A71, Ser A73, Arg A 86, Ser C147, Leu A148, and Thr C149 were the residues reported for UBE2Q1.
Molecular Docking
OC proteins were docked against the phytochemical ligand database. A strict filter that included four factors was used to rank docked compounds: hydrogen bonding strength, maximum binding pocket occupancy with lowest Gibbs free energy, and other potential non-covalent interactions, all calculated and represented using an S-score function. Top docking poses were chosen from among 10,326 docked molecules. The ranking criteria included threshold-based criteria that required a ligand to exhibit the desired S-score values (stronger the affinities and interaction, lower the score) and bind to the target OC proteins using all of the hotspot conserved residues in the binding pocket (Table 1).
Table 1. Docking statistics of target proteins against plant compounds
|
Compounds I’D |
Compounds Name |
Binding Affinity |
RMSD |
Interacting residues |
|
NY-ESO-1 |
||||
|
5281520 |
Humulene |
-17.65 kcal/mol |
1.54 |
Asp A30, Thr A31, Gln A32, Leu B65, Ser B52, Tyr A27, Tyr B63 |
|
1130 |
Thiamine |
-15.34 kcal/mol |
0.87 |
Tyr A27, Ser B55, Asp B53, Ser B52, Tyr B63, Phe A241, Gln A32 |
|
6537501 |
Deoxyactein |
-14.76 kcal/mol |
1.21 |
Tyr A27, Gln A32, Arg A48, Tyr B67, Ser B52, His B51, Asp A30 |
|
RUNX3 |
||||
|
5324208 |
(3-(Benzyloxy)isoxazol-5-yl)methanol |
-16.65 kcal/mol |
0.77 |
Tyr C27, Ser D55, Tyr D63, Phe C241, Asp D53, Ser D52, Gln C32, Leu D65, Tyr D26 |
|
5352042 |
Buddledin C |
-15.76 kcal/mol |
1.25 |
Tyr D26, Thr C233, Ala C211, Tyr D63, Pro C235 |
|
65067 |
Terthiophene |
-13.87 kcal/mol |
1.74 |
Arg C48, Tyr D67, Phe C241, Tyr C27 |
|
UBE2Q1 |
||||
|
444 |
Bupropion |
-14.65 Kcal/mol |
1.65 |
Asp A38, Val A32, Leu A148, Ser A73, Asn A71, Arg A 86 |
|
838 |
DL-Adrenaline |
-12.23 Kcal/mol |
1.98 |
Lys A75, Asp A38, Ser A73, Ser C147, Thr C149 |
|
10550692 |
Vibsanin H |
-11.93 Kcal/mol |
2.71 |
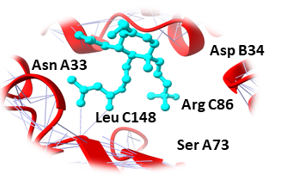
Ser A73, Leu C148, Asp B34, Asn A33, Arg C86 |
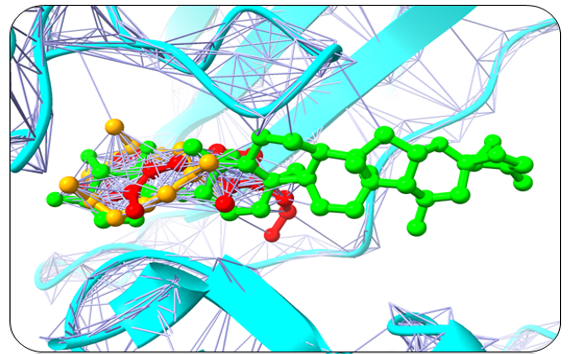
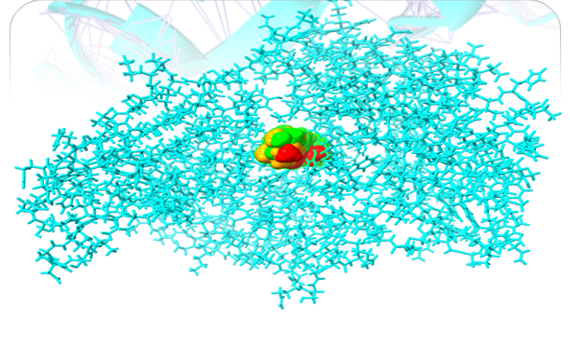
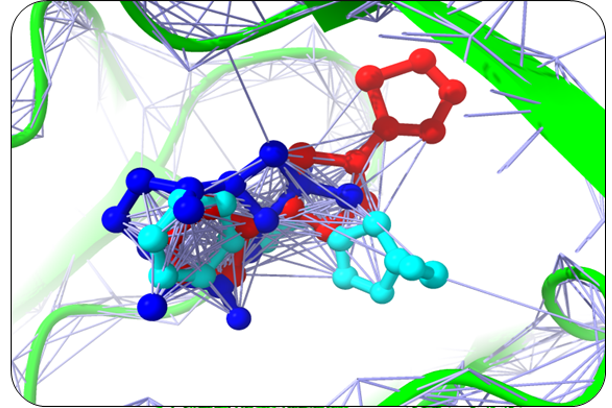
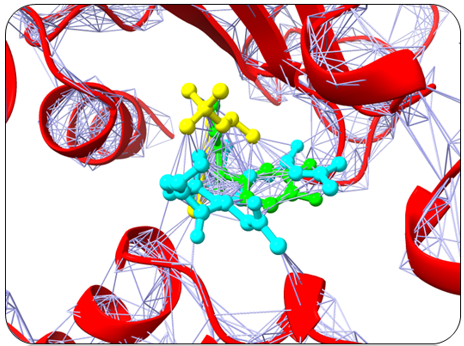
Humulene, Thiamine, and Deoxyactein were found to bind with high affinity within the active sites of the NY-ESO-1 (Figure 1). Humulene was bound to NY-ESO-1 protein, having a score of -17.65 KJ / mol creating hydrogen bonds with Asp A30, Thr A31, Gln A32, Leu B65, Ser B52, Tyr A27, and Tyr B63 side chains (Figure 1a) and Thiamine D was bound having a binding score of -15.34 kcal/mol creating hydrogen bonds with Tyr A27, Ser B55, Asp B53, Ser B52, Tyr B63, Phe A241, and Gln A32 side chains (Figure 1b). In contrast, Deoxyactei was bound to NY-ESO-1 protein with a -14.76 KJ / mol, forming hydrogen bonds with the side chains of Tyr A27, Gln A32, Arg A48, Tyr B67, Ser B52, His B51, and Asp A30 (Figure 1c).
|
|
|
|
a) |
|
|
|
|
|
b) |
|
|
|
|
|
c) |
|
|
Figure 1. Interaction mechanisms and binding modes of new NY-ESO-1 protein inhibitors Interaction analysis of Humulene (a), Thiamine (b), and Deoxyactein (c) |
|
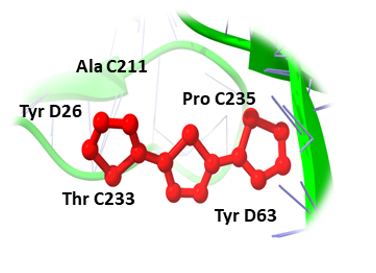
Likewise, (3-(Benzyloxy) isoxazol-5-yl) methanol, Buddledin C, and Terthiophene were observed to have a high affinity within the active sites of the RUNX3 (Figure 2). (3-(Benzyloxy) isoxazol-5-yl) methanol was bound to RUNX3 protein, having a score of -16.65 KJ / mol creating hydrogen bonds with the side chains of Tyr C27, Ser D55, Tyr D63, Phe C241, Asp D53, Ser D52, Gln C32, Leu D65, and Tyr D26 (Figure 2a) and Buddledin C was bound with a binding score of -15.76 kcal/mol creating hydrogen bonds with side chains of Tyr D26, Thr C233, Ala C211, Tyr D63, and Pro C235 (Figure 2b). While Terthiophene was bound to RUNX3 protein with a -13.87 KJ / mol score, creating hydrogen bonds with the side chains of Arg C48, Tyr D67, Phe C241 and Tyr C27 (Figure 2c).
|
|
|
|
a) |
|
|
|
|
|
b) |
|
|
|
|
|
c) |
|
|
Figure 2. Interaction mechanisms and binding modes of new RUNX3 protein inhibitors. Interaction analysis of (3-(Benzyloxy) isoxazol-5-yl) methanol (a), Buddledin C (b), and Terthiophene (c) |
|
Similarly, Bupropion, DL-Adrenaline, and Vibsanin H were found to bind with high affinity within the active sites of the UBE2Q1 (Figure 3). Bupropion was bound to UBE2Q1 protein with a score of -14.65 KJ / mol creating hydrogen bonds with the side chains of Asp A38, Val A32, Leu A148, Ser A73, Asn A71, and Arg A 86 (Figure 3a), and DL-Adrenaline was bound with a binding score of -12.23 kcal/mol creating hydrogen bonds with side chains of Lys A75, Asp A38, Ser A73, Ser C147, and Thr C149 (Figure 3b). Vibsanin H was bound to UBE2Q1 protein with a -11.93 KJ / mol creating hydrogen bonds with the Ser A73, Leu C148, Asp B34, Asn A33, and Arg C86 side chains (Figure 3c).
|
|
|
|
a) |
|
|
|
|
|
b) |
|
|
|
|
|
c) |
|
|
Figure 3. Interaction mechanisms and binding modes of new UBE2Q1 protein inhibitors. Interaction analysis of Bupropion (a), DL-Adrenaline (b), and Vibsanin H (c) |
|
Drug Scan/ADMET Results
The drug-likeness of chosen compounds was anticipated using the Molinspiration server, which relied on the Lipinski Rules of Five. The chosen candidates had no violations of the "rule of five" and expressed drug-like properties.
For further validation of the drug likeliness potential, pharmacokinetic properties of all the candidate compounds were assessed using the admetSAR server, and their results are indicated in Table 2.
Table 2. ADMET profiling of best docked compounds
|
Target Receptors |
NY-ESO-1 |
RUNX3 |
UBE2Q1 |
||||||
|
Compounds I’Ds |
5281520 |
1130 |
6537501 |
5324208 |
5352042 |
65067 |
444 |
838 |
10550692 |
|
Absorption |
|||||||||
|
Human Intestinal Absorption |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
|
Blood-Brain Barrier |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
|
Caco-2 Permeable |
+ |
- |
- |
- |
+ |
- |
- |
- |
+ |
|
Distribution |
|||||||||
|
P-Glycoprotein Substrate |
Substrate |
Substrate |
Substrate |
Substrate |
Substrate |
Substrate |
Substrate |
Substrate |
Substrate |
|
P-Glycoprotein Inhibitor I |
Inhibitor |
Non-Inhibitor |
Inhibitor |
Inhibitor |
Non-Inhibitor |
Non-Inhibitor |
Non-Inhibitor |
Non-Inhibitor |
Non-Inhibitor |
|
Metabolism |
|||||||||
|
CYP450 2C9 Substrate |
Substrate |
Non-Substrate |
Substrate |
Non-Substrate |
Substrate |
Non-Substrate |
Non-Substrate |
Substrate |
Non-Substrate |
|
CYP450 2D6 Substrate |
Substrate |
Non-Substrate |
Substrate |
Substrate |
Non-Substrate |
Non-Substrate |
Non-Substrate |
Non-Substrate |
Non-Substrate |
|
CYP450 3A4 Substrate |
Substrate |
Non-Substrate |
Substrate |
Substrate |
Non-Substrate |
Substrate |
Non-Substrate |
Non-Substrate |
Substrate |
|
CYP450 1A2 Substrate |
Non-Substrate |
Non-Substrate |
Substrate |
Non-Substrate |
Non-Substrate |
Non-Substrate |
Substrate |
Non-Substrate |
Non-Substrate |
|
CYP450 2C9 Inhibitor |
Non-Inhibitor |
Non-Inhibitor |
Non-Inhibitor |
Non- Inhibitor |
Inhibitor |
Non-Inhibitor |
Non-Inhibitor |
Non-Inhibitor |
Non-Inhibitor |
|
CYP450 2D6 Inhibitor |
Inhibitor |
Non-Inhibitor |
Inhibitor |
Non- Inhibitor |
Non-Inhibitor |
Non-Inhibitor |
Inhibitor |
Non-Inhibitor |
Non-Inhibitor |
|
CYP450 2C19 Inhibitor |
Non-Inhibitor |
Inhibitor |
Non-Inhibitor |
Inhibitor |
Non-Inhibitor |
Inhibitor |
Non-Inhibitor |
Non-Inhibitor |
Non-Inhibitor |
|
CYP450 3A4 Inhibitor |
Non-Inhibitor |
Non-Inhibitor |
Non-Inhibitor |
Non-Inhibitor |
Inhibitor |
Non-Inhibitor |
Non-Inhibitor |
Non-Inhibitor |
Inhibitor |
|
CYP450 Inhibitory Promiscuity |
Low CYP Inhibitory Promiscuity |
Low CYP Inhibitory Promiscuity |
Low CYP Inhibitory Promiscuity |
Low CYP Inhibitory Promiscuity |
Low CYP Inhibitory Promiscuity |
Low CYP Inhibitory Promiscuity |
Low CYP Inhibitory Promiscuity |
Low CYP Inhibitory Promiscuity |
Low CYP Inhibitory Promiscuity |
|
Toxicity |
|||||||||
|
Ames Toxicity |
Non-Toxic |
Non-Toxic |
Non-Toxic |
Non-Toxic |
Non-Toxic |
Non-Toxic |
Non-Toxic |
Non-Toxic |
Non-Toxic |
|
Carcinogenicity |
Non-Carcinogens |
Non-Carcinogens |
Non-Carcinogens |
Non-Carcinogens |
Non-Carcinogens |
Non-Carcinogens |
Non-Carcinogens |
Non-Carcinogens |
Non-Carcinogens |
Binding Free Energy Calculations
Binding free energies were estimated by employing MMGBSA/MMPBSA methods to better understand the complexes' binding ability with OC proteins. Stable complexes are generated because all of the binding interactions are energetically favorable. In all of the complexes, gas-phase energy dominates the system energy, with van der Waals playing an important role, and electrostatic energy has a minor role. In binding, the polar solvation energy is unfavorable, whereas, in complex equilibration, the nonpolar energy appears to be favorable. NY-ESO-1 receptor showed the total energy of -30.35 kcal/mol (5281520), -36.54 kcal/mol (1130), -41.54 kcal/mol (6537501). Although the receptor RUNX3 showed delta total as -29.74 kcal/mol (5324208), -24.58 kcal/mol (5352042), -23.12 kcal/mol (65067) for all the complexes meanwhile the receptor UBE2Q1 showed -21.58 kcal/mol (444), -30.52 kcal/mol (838), and for last compound it was found to be -27.04 kcal/mol (10550692).
Molecular Dynamic Simulation
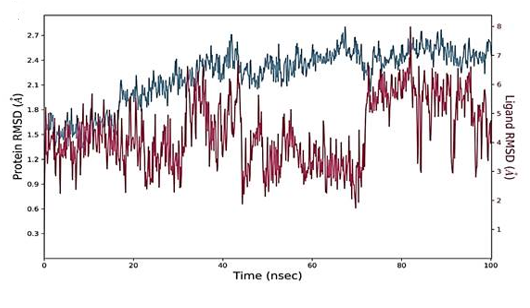
To gain a better understanding of targets' dynamics in the presence of screened hits, a 100-ns molecular dynamic simulation was run. The structural stability of docked complexes was confirmed using statistical characteristics like RMSD, RMSF, and interactions. The carbon alpha atoms' Root Mean Square Deviations (RMSD) was examined first.
Root Mean Square Deviations (RMSD) and Root Mean Square Fluctuations (RMSF)
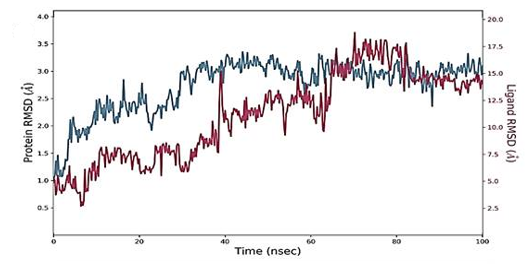
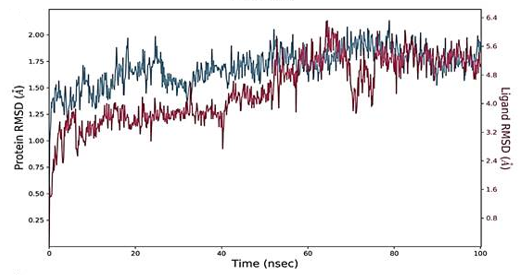
The RMSD figure deviates from the initial docked complex intermolecular conformation, indicating structural changes. As the simulation time progresses, a uniform RMSD plot indicates system structural equilibrium and increased intermolecular strength (Figure 4). During the initial 60 ns of simulation time, the Humulene/ NYESO-1complex showed a minor deviation of 0.5 Ao after that complex remained stable, as shown in Figure 4a. The second (3-(Benzyloxy) isoxazol-5-yl) methanol/ RUNX3 complex showed minor deviations on its first jump up to 50 ns before achieving stability, as illustrated in Figure 4b. The third Bupropion/ UBE2Q1 complex showed a minor deviation of 1 Ao to 1.5 Ao between 40 to 70 ns other than it remained stable, as indicated in Figure 4c.
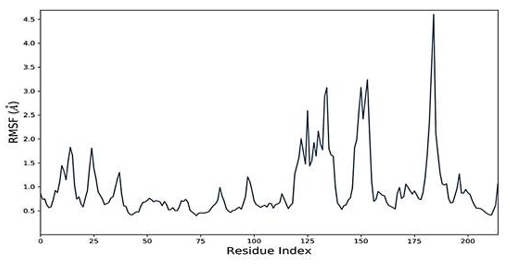
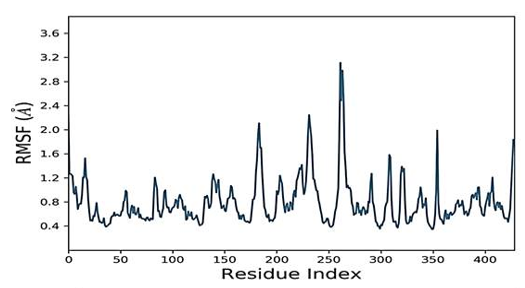
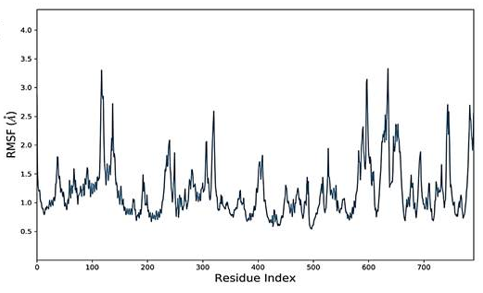
The simulated complexes' Root Mean Square Fluctuations (RMSF) was computed. RMSF analysis helps understand how these fluctuations affect complex stability and identify flexible residues in targeted proteins (Figure 4). Graphs of the Humulene/ NYESO-1 complex indicate minor fluctuations at residue number 180, as indicated in Figure 4e. In contrast, the second (3-(Benzyloxy) isoxazol-5-yl) methanol/ RUNX3 complex and third Bupropion/ UBE2Q1 complex showed a deviation at one point up to the residue number 250 for a second, as shown in Figures 4f and 4g.
|
|
|
a) |
|
|
|
b) |
|
|
|
c) |
|
|
|
e) |
|
|
|
f) |
|
|
|
g) |
|
Figure 4. a) RMSD analysis of Humulene/ NYESO-1complex b) RMSD analysis of (3-(Benzyloxy) isoxazol-5-yl) methanol/ RUNX3 complex c) RMSD analysis of Bupropion/ UBE2Q1 complex e). RMSF analysis of Humulene/ NYESO-1complex f) RMSF analysis of (3-(Benzyloxy)isoxazol-5-yl) methanol/ RUNX3 complex g) RMSF analysis of Bupropion/ UBE2Q1 complex |
Radius of Gyration (RoG) & Solvent Accessible Surface Area (SASA)
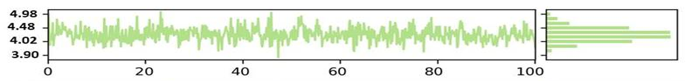
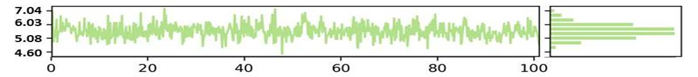
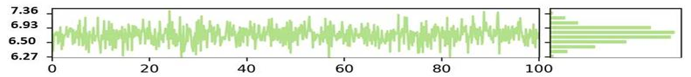
The NYESO-1 target Radius of gyration (RoG) plot trajectories for the first complex (Humulene/ NYESO-1) exhibited good stability throughout the 100 ns period between 4.02 to 4.48 Ao shown in the Figure 5a. However, the second complex second (3-(Benzyloxy) isoxazol-5-yl)methanol/ RUNX3 complex showed stability trajectories between 5.08 Angstrom to 6.03Ao (Figure 5b). The third Bupropion/ UBE2Q1 complex showed stability throughout 100ns at 6.85 Ao, as indicated in Figure 5c.
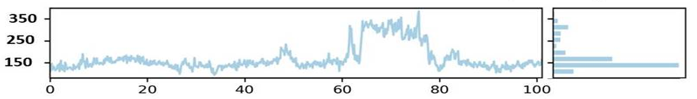
SASA is an alternative method for maintaining protein stability and folding. The calculated SASA values are displayed in Figure 5. The average SASA values for the first complex Humulene/ NYESO-1 were 150 Å2 for the first complex, with a minor deviation from 60ns to 80ns Å2 (Figure 5d). The second complex (3-(Benzyloxy) isoxazol-5-yl) methanol/ RUNX3 showed stability up to 50ns at 130 Å2. After a minor deviation, it returns to stability at 130Å2, indicating no significant differences in all systems' available areas throughout the simulation process, as shown in Figure 5e. Although the 3rd complex Bupropion/ UBE2Q1 showed stability up to the residue number 230 Å2 throughout 100 ns (Figure 5f).
|
Radius of gyration (RoG) |
|
|
|
a) |
|
|
|
b) |
|
|
|
c) |
|
Solvent accessible surface area (SASA) |
|
|
|
d) |
|
|
|
e) |
|
|
|
f) |
|
Figure 5. a) RoG analysis of Humulene/ NYESO-1complex. b) RoG analysis of (3-(Benzyloxy) isoxazol-5-yl) methanol/ RUNX3 complex c) RoG analysis of Bupropion/ UBE2Q1 complex. d) SASA analysis of Humulene/ NYESO-1complex. e) SASA analysis of (3-(Benzyloxy) isoxazol-5-yl) methanol/ RUNX3 complex. f) SASA analysis of Bupropion/ UBE2Q1 complex |
OC remains the deadliest gynecologic cancer. The mortality rate of OC is expected to increase significantly by 2022 (Bray et al., 2020). Several factors contribute to the higher mortality rate of OC, including the slow progression of disease symptoms, stealthy and asymptomatic progression of malignant tumorous cells, and the lack of relevant methods for cancer diagnosis even in advanced stages. This is why the OC is known as the "Silent Killer" (Yousefi et al., 2017; Danilova et al., 2020; Henriksen et al., 2020; Nakagawa & Yamashita, 2022). The treatment is far from ideal; conventional treatment strategies such as radiotherapy, chemotherapy, and surgery have been used in the past; however, these options have significant drawbacks and are often unproductive (Henriksen et al., 2020).
Researchers studied this cancer type and discovered high-risk genes, and inhibiting these target proteins will help us stop cancer progression. In silico studies make it easier to screen ligands and analyze their interactions with target proteins. Our study aims to compare and identify the most appropriate compound that binds to our target protein and reduces its tumor-causing actions, thereby inhibiting tumor growth (Barua et al., 2018). Our target proteins, in this case, are NY-ESO-1, RUNX3, and UBE2Q1.
A famous cancer-testis antigen (CTA) re-expressed in various cancers is the New York esophageal squamous cell carcinoma 1 (NY-ESO-1). It is a promising candidate target for cancer immunotherapy due to its ability to induce humoral immune responses and spontaneous cellular and its limited expression pattern (Thomas et al., 2018). RUNX3, a Runt domain transcription factor, is found in the nuclei of OC cell lines and plays an oncogenic role in the disease. RUNX3 could be used as a marker for OC diagnosis or as a potential therapeutic target in conjunction with other known targets. UBE2Q1 (Ubiquitin Conjugating Enzyme E2 Q1) is a potential predictive and prognostic marker for relapse-free survival in patients with serous OC (Topno et al., 2021; Nurcahyo et al., 2022).
To treat various diseases for centuries, plants have been utilized. Lately, there has been an increasing emphasis on identifying and using plant-derived compounds that can act as potent anti-cancer agents (Barua et al., 2018). Hence, 2500 natural, plant-derived compounds were selected and tested for inhibitory properties in our study.
Molecular docking has been widely used to investigate plant compound binding interactions with target proteins' active sites. Hierarchical virtual screening approaches have already recognized potential anti-cancer compounds against various cancers in anti-cancer drug discovery (Awasthi et al., 2015; Wang et al., 2016; Gogoi et al., 2017; Yousuf et al., 2017; Kumar et al., 2019; Rehan & Bajouh, 2019; Opo et al., 2021). This study docked OC proteins (NY-ESO-1, RUNX3, and UBE2Q1) against the phytochemical ligand database. Molecular docking is a technique for predicting how compounds bind to their protein targets. These computational approaches provide information on compounds' activity and binding affinity for their target protein.
Humulene, Thiamine, Deoxyactein, (3-(Benzyloxy) isoxazol-5-yl) methanol, Buddledin C, Terthiophene, Bupropion, DL-Adrenaline, and Vibsanin H were discovered to bind with high affinity within the active sites of the targeted OC proteins. The discovered chemicals may have additive or synergistic effects against OC. By efficiently hindering and targeting the catalytic function of target proteins, these inhibitors could lead to a single anti-OC therapeutic solution.
These nine compounds were chosen for additional computational investigations to better understand their molecular interaction mechanisms, binding modes, and ADMET evaluation. The ADMET properties of compounds expected to be drugs are being studied further. A major challenge in the drug development process is determining the ADMET properties of compounds. Toxicity and poor pharmacokinetic properties account for most drugs that fail to pass the drug approval process. The development of high-performance and quick ADMET profiling tests has helped detect active compounds during the early stages of drug discovery.
The Blood-brain Barrier (BBB) permeation and Gastrointestinal absorption (GI) of drug molecules indicate drug distribution and absorption. Caco-2 permeability further demonstrated the compounds' absorption (Abdelrheem et al., 2021). Additionally, several cytochromes (CYPs) regulate drug metabolism, with CYP2D6, CYP3A4, CYP2C19, CYP2C9, and CYP1A2 required for drug molecule biotransformation (Das et al., 2021). Furthermore, p-glycoprotein inhibitors decrease the bioavailability of the drug known to be transported by it. A toxicity prediction study was carried out to evaluate the compounds' safety profile (Mohammed, 2021). All of the compounds chosen were discovered to be non-toxic and non-carcinogenic. These results imply that certain compounds could be developed as drugs and used to treat OC.
MD simulations are a viable method for investigating the underlying dynamics of protein-ligand interactions. MD simulations and MMGBSA/MMPBSA analysis were studied on the best-docked complexes with Humulene, (3-(Benzyloxy) isoxazol-5-yl) methanol, and Bupropion inhibitors since these ligands represented high binding affinity, as evidenced by a good molecular interaction network and a high dock score. MD simulation and MMGBSA/MMPBSA studies revealed that these compounds were stable as potent inhibitors within the protein binding pocket. These inhibitors may give rise to a single therapeutic solution against the OC by effectively inhibiting and targeting the catalytic function of three target proteins. Hence, our findings regarding the bioactivity of Humulene, Thiamine, Deoxyactein, (3-(Benzyloxy) isoxazol-5-yl) methanol, Buddledin C, Terthiophene, Bupropion, DL-Adrenaline, and Vibsanin H warrant additional research for structure-based lead optimization.
Conclusion
Natural compounds have always been a rich source of active compounds with a wide range of structures; hence, these compounds have served as a special source of inspiration for medicinal chemists. To design therapeutic interventions, the current study aimed to conduct molecular dynamics and molecular docking simulation studies on natural compounds targeting ovarian cancer proteins NY-ESO-1, RUNX3, and UBE2Q1. We discussed computational validations here, but in vivo studies or experimental evaluations are recommended for further research. Several of these compounds have the potential to be the next breakthroughs in ovarian cancer early detection.
Acknowledgments: None
Conflict of interest: None
Financial support: None
Ethics statement: None
Abdelrheem, D. A., Rahman, A. A., Elsayed, K. N., Abd El-Mageed, H. R., Mohamed, H. S., & Ahmed, S. A. (2021). Isolation, characterization, in vitro anticancer activity, dft calculations, molecular docking, bioactivity score, drug-likeness and admet studies of eight phytoconstituents from brown alga sargassum platycarpum. Journal of Molecular Structure, 1225, 129245.
Alhazmi, R. A., Khayat, S. K., Albakri, M. H., Alruwaili, W. S., Bayazed, H. A., Almubarak, S. A., Albahrani, A. A., Alshahrani, A. A., Alharkan, A. A., Alreqei, H. M., et al. (2022). An Overview on the Assessment and Management of Polycystic Ovarian Syndrome. World Journal of Environmental Biosciences, 11(1), 17-23. doi:10.51847/Yaaa2745ZY
Alhumaydhi, F. A., Rauf, A., Rashid, U., Bawazeer, S., Khan, K., Mubarak, M. S., Aljohani, A. S., Khan, H., El-Saber Batiha, G., El-Esawi, M. A., et al. (2021). In Vivo and In Silico Studies of Flavonoids Isolated from Pistacia integerrima as Potential Antidiarrheal Agents. ACS Omega, 6(24), 15617-15624.
AlHussain, B. S., AlFantoukh, M. A. M., Alasmari, K. M. A., AlHrab, F. A., Alotaibi, F. A., Alaybani, W. H., & AlOtaibi, I. A. B. (2022). Clinical Knowledge of Orthodontics Complication and Emergencies Among Interns and Dentists in Riyadh City. Annals of Dental Specialty, 10(2), 45-51. doi:10.51847/9H6hhUJo8E
AlShehri, O. M., Jali, N. M., Almutairi, Y. M., Aljrais, M. M., Alsirhani, A. M., & AlQudairi, A. S. (2022). Common Causes of Cusp Fracture in Adults; A Systematic Review. Annals of Dental Specialty, 10(2), 35-38. doi:10.51847/jBTdUqt7Cx
Auner, V., Sehouli, J., Oskay-Oezcelik, G., Horvat, R., Speiser, P., & Zeillinger, R. (2010). ABC transporter gene expression in benign and malignant ovarian tissue. Gynecologic Oncology, 117(2), 198-201.
Awasthi, M., Singh, S., Pandey, V. P., & Dwivedi, U. N. (2015). Molecular docking and 3D-QSAR-based virtual screening of flavonoids as potential aromatase inhibitors against estrogen-dependent breast cancer. Journal of Biomolecular Structure and Dynamics, 33(4), 804-819.
Barua, A., Kesavan, K., & Jayanthi, S. (2018). Molecular Docking Studies of Plant Compounds to Identify Efficient Inhibitors for Ovarian Cancer. Research Journal of Pharmacy and Technology, 11(9), 3811-3815.
Bittrich, S., Rose, Y., Segura, J., Lowe, R., Westbrook, J. D., Duarte, J. M., & Burley, S. K. (2022). RCSB Protein Data Bank: improved annotation, search and visualization of membrane protein structures archived in the PDB. Bioinformatics, 38(5), 1452-1454.
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., & Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians, 68(6), 394-424.
Burlaka, I. (2022). Approach to correction of apoptotic disorders in children with early diabetic nephropathy. Journal of Advanced Pharmacy Education and Research, 12(2), 104-109. doi:10.51847/G6i3231Jnm
Cheng, F., Li, W., Zhou, Y., Shen, J., Wu, Z., Liu, G., Lee, P. W., & Tang, Y. (2012). admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. ACS Publications.
Danilova, A., Misyurin, V., Novik, A., Girdyuk, D., Avdonkina, N., Nekhaeva, T., Emelyanova, N., Pipia, N., Misyurin, A., & Baldueva, I. (2020). Cancer/testis antigens expression during cultivation of melanoma and soft tissue sarcoma cells. Clinical Sarcoma Research, 10(1), 1-14.
Das, P., Majumder, R., Mandal, M., & Basak, P. (2021). In-Silico approach for identification of effective and stable inhibitors for COVID-19 main protease (Mpro) from flavonoid based phytochemical constituents of Calendula officinalis. Journal of Biomolecular Structure and Dynamics, 39(16), 6265-6280.
de Vries, S. J., & Bonvin, A. M. (2011). CPORT: a consensus interface predictor and its performance in prediction-driven docking with HADDOCK. PloS one, 6(3), e17695.
Devi, K. P., Rajavel, T., Habtemariam, S., Nabavi, S. F., & Nabavi, S. M. (2015). Molecular mechanisms underlying anticancer effects of myricetin. Life Sciences, 142, 19-25.
Ghilardi, C., Moreira Barbosa, C., Brunelli, L., Ostano, P., Panini, N., Lupi, M., Anastasia, A., Fiordaliso, F., Salio, M., Formenti, L., et al. (2022). PGC1α/β expression predicts therapeutic response to oxidative phosphorylation inhibition in ovarian canceroxphos inhibition as therapeutic option in ovarian cancer. Cancer Research.
Gogoi, B., Gogoi, D., Silla, Y., Kakoti, B. B., & Bhau, B. S. (2017). Network pharmacology-based virtual screening of natural products from Clerodendrum species for identification of novel anti-cancer therapeutics. Molecular BioSystems, 13(2), 406-416.
Gupta, S., Afaq, F., & Mukhtar, H. (2001). Selective growth-inhibitory, cell-cycle deregulatory and apoptotic response of apigenin in normal versus human prostate carcinoma cells. Biochemical and Biophysical Research Communications, 287(4), 914-920.
Haque, A., Baig, G. A., Alshawli, A. S., Sait, K. H. W., Hafeez, B. B., Tripathi, M. K., Alghamdi, B. S., Mohammed Ali, H. S., & Rasool, M. (2022). Interaction Analysis of MRP1 with Anticancer Drugs Used in Ovarian Cancer: In Silico Approach. Life, 12(3), 383.
Heimes, D., Mark, N. A., Kuchen, R., Pabst, A., Becker, P., Kyyak, S., Thiem, D. G. E., Schulze, R., & Kämmerer, P. W. (2022). Awareness and Preparedness of Saudi Dental Trainees on Medication-Related Osteonecrosis of the Jaw. Annals Journal of Dental and Medical Assisting, 2(2), 1-9. doi:10.51847/SkHmFpUrKD
Henriksen, J. R., Donskov, F., Waldstrøm, M., Jakobsen, A., Hjortkjaer, M., Petersen, C. B., & Dahl Steffensen, K. (2020). Favorable prognostic impact of Natural Killer cells and T cells in high-grade serous ovarian carcinoma. Acta Oncologica, 59(6), 652-659.
Ikot, A. N., Okorie, U. S., Osobonye, G., Amadi, P. O., Edet, C. O., Sithole, M. J., Rampho, G. J., & Sever, R. (2020). Superstatistics of Schrödinger equation with pseudo-harmonic potential in external magnetic and Aharanov-Bohm fields. Heliyon, 6(4), e03738.
Irwin, J. J., & Shoichet, B. K. (2005). ZINC− a free database of commercially available compounds for virtual screening. Journal of chemical Information and Modeling, 45(1), 177-182.
Kabir, M., Nantasenamat, C., Kanthawong, S., Charoenkwan, P., & Shoombuatong, W. (2022). Large-scale comparative review and assessment of computational methods for phage virion proteins identification. EXCLI Journal, 21, 11-29.
Kbirou, A., Sayah, M., Sounni, F., Zamd, M., Benghanem, M. G., Dakir, M., Debbagh, A., & Aboutaib, R. (2022). Obstructive oligo-anuria revealing pelvic gynecological cancers, analysis of a series of 102 cases. Annals of Medicine and Surgery, 75, 103332.
Khalifa, I., Zhu, W., Mohammed, H. H. H., Dutta, K., & Li, C. (2020). Tannins inhibit SARS‐CoV‐2 through binding with catalytic dyad residues of 3CLpro: An in silico approach with 19 structural different hydrolysable tannins. Journal of Food Biochemistry, 44(10), e13432.
Kim, S., Thiessen, P. A., Bolton, E. E., Chen, J., Fu, G., Gindulyte, A., Han, L., He, J., He, S., Shoemaker, B. A., et al. (2016). PubChem substance and compound databases. Nucleic Acids Research, 44(D1), D1202-D1213.
Kumar D, Gurunathan D, Jabin Z, Talal S. Comparative Efficacy of Aromatherapy and Conscious Sedation in Pediatric Dental Anxiety Management. Ann J Dent Med Assist. 2022;2(1):14-21. doi:10.51847/TVRFhVaiIQ
Kumar, A., Rathi, E., & Kini, S. G. (2019). E-pharmacophore modelling, virtual screening, molecular dynamics simulations and in-silico ADME analysis for identification of potential E6 inhibitors against cervical cancer. Journal of Molecular Structure, 1189, 299-306.
Ledermann, J. A., Raja, F. A., Fotopoulou, C., Gonzalez-Martin, A., Colombo, N., & Sessa, C. (2013). Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology, 24, vi24-vi32.
Li, Y., Zhang, S., Bao, Z., Sun, N., & Lin, S. (2022). Explore the activation mechanism of alcalase activity with pulsed electric field treatment: Effects on enzyme activity, spatial conformation, molecular dynamics simulation and molecular docking parameters. Innovative Food Science & Emerging Technologies, 102918.
Manchanda, R. (2022). Special Issue “Gynaecological Cancers Risk: Breast Cancer, Ovarian Cancer and Endometrial Cancer”. Cancers, 14(2), 319.
Mishra, A., & Singh, A. (2022). Discovery of Histone Deacetylase Inhibitor Using Molecular Modeling and Free Energy Calculations. ACS Omega.
Mohammed, I. (2021). Virtual screening of Microalgal compounds as potential inhibitors of Type 2 Human Transmembrane serine protease (TMPRSS2). arXiv preprint arXiv:2108.13764.
Mumtaz, A., Ashfaq, U. A., ul Qamar, M. T., Anwar, F., Gulzar, F., Ali, M. A., Saari, N., & Pervez, M. T. (2017). MPD3: a useful medicinal plants database for drug designing. Natural Product Research, 31(11), 1228-1236.
Nakagawa, N., & Yamashita, T. (2022). Comparative study of traditional face-to-face teaching, audience response system, and a flipped classroom plus audience response. Journal of Advanced Pharmacy Education and Research, 12(1), 9-16. doi:10.51847/ODLhsGztZ9
Nurcahyo, H., Sumiwi, S. A., Halimah, E., & Wilar, G. (2022). Secondary metabolitm determination from Brebes shallot’s ethanol extract and its ethyl acetate fraction “Allium ascalonicum L.”. Journal of Advanced Pharmacy Education and Research, 12(1), 70-73. doi:10.51847/NfNMFJB9ac
Opo, F. A., Rahman, M. M., Ahammad, F., Ahmed, I., Bhuiyan, M. A., & Asiri, A. M. (2021). Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein. Scientific Reports, 11(1), 1-17.
Pahwa, R., Chhabra, J., Kumar, R., & Narang, R. (2022). Melphalan: Recent insights on synthetic, analytical and medicinal aspects. European Journal of Medicinal Chemistry, 114494.
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., & Ferrin, T. E. (2004). UCSF Chimera—a visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25(13), 1605-1612.
Pettersen, E. F., Goddard, T. D., Huang, C. C., Meng, E. C., Couch, G. S., Croll, T. I., Morris, J. H., & Ferrin, T. E. (2021). UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Science, 30(1), 70-82.
Podvinec, M., Lim, S. P., Schmidt, T., Scarsi, M., Wen, D., Sonntag, L. S., Sanschagrin, P., Shenkin, P. S., & Schwede, T. (2010). Novel inhibitors of dengue virus methyltransferase: discovery by in vitro-driven virtual screening on a desktop computer grid. Journal of Medicinal Chemistry, 53(4), 1483-1495.
Rehan, M., & Bajouh, O. S. (2019). Virtual screening of naphthoquinone analogs for potent inhibitors against the cancer‐signaling PI3K/AKT/mTOR pathway. Journal of Cellular Biochemistry, 120(2), 1328-1339.
Release, S. (2017). 3: Desmond molecular dynamics system, DE Shaw research, New York, NY, 2017. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY.
Remizova, A. A., Bitarov, P. A., Epkhiev, A. A., & Remizov, N. O. (2022). Reparative-regenerative features of bone tissue in experimental animals treated with titanium implants. Journal of Advanced Pharmacy Education and Research, 12(2), 110-116. doi:10.51847/Sprxb1DKyv
Roy, P., Sur, S., Das, S., & Wui, W. T. (2022). Phytochemical-conjugated bio-safe gold nanoparticles in breast cancer: a comprehensive update. Breast Cancer, 1-17.
Siegel, R. L., Miller, K. D., & Jemal, A. (2016). Cancer statistics, 2016. CA: a cancer journal for clinicians, 66(1), 7-30.
Tavsan, Z., & Kayali, H. A. (2019). Flavonoids showed anticancer effects on the ovarian cancer cells: Involvement of reactive oxygen species, apoptosis, cell cycle and invasion. Biomedicine & Pharmacotherapy, 116, 109004.
Thomas, R., Al-Khadairi, G., Roelands, J., Hendrickx, W., Dermime, S., Bedognetti, D., & Decock, J. (2018). NY-ESO-1 based immunotherapy of cancer: current perspectives. Frontiers in Immunology, 9, 947.
Topno, R., Singh, I., Kumar, M., & Agarwal, P. (2021). Integrated bioinformatic analysis identifies UBE2Q1 as a potential prognostic marker for high grade serous ovarian cancer. BMC Cancer, 21(1), 1-13.
Torre, L. A., Trabert, B., DeSantis, C. E., Miller, K. D., Samimi, G., Runowicz, C. D., Gaudet, M. M., Jemal, A.., & Siegel, R. L. (2018). Ovarian cancer statistics, 2018. CA: a Cancer Journal for Clinicians, 68(4), 284-296.
Vilar, S., Cozza, G., & Moro, S. (2008). Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Current Topics in Medicinal Chemistry, 8(18), 1555-1572.
Wang, L., Chen, L., Yu, M., Xu, L. H., Cheng, B., Lin, Y. S., Gu, Q., He, X. H., & Xu, J. (2016). Discovering new mTOR inhibitors for cancer treatment through virtual screening methods and in vitro assays. Scientific Reports, 6(1), 1-13.
Wang, Y. T., Yang, Z. X., Piao, Z. H., Xu, X. J., Yu, J. H., & Zhang, Y. H. (2021). Prediction of flavor and retention index for compounds in beer depending on molecular structure using a machine learning method. RSC Advances, 11(58), 36942-36950.
Yousefi, H., Yuan, J., Keshavarz-Fathi, M., Murphy, J. F., & Rezaei, N. (2017). Immunotherapy of cancers comes of age. Expert Review of Clinical Immunology, 13(10), 1001-1015.
Yousuf, Z., Iman, K., Iftikhar, N., & Mirza, M. U. (2017). Structure-based virtual screening and molecular docking for the identification of potential multi-targeted inhibitors against breast cancer. Breast Cancer: Targets and Therapy, 9, 447.