Flavone Derivatives as Potential Inhibitors of SARS-Cov-2rdrp through Computational Studies
|
Mohammed Merzouki, Lamiae Bourassi, Rania Abidi, Boufelja Bouammali, Allal Challioui* Laboratory of Applied Chemistry and Environment (LCAE-ECOMP), Faculty of Sciences, University Mohamed Premier, PB 60000, Oujda, Morocco.
Ayoub Bekkouch Biology and Health Laboratory (BHL), Faculty of Ibn Tofail University, Kenitra Morocco.
Raed Alkowni Department of Biology and Biotechnology, An-Najah National University, Nablus, Palestine.
Belkheir Hammouti Laboratory of Industrial, Engineering, Energy and Environment (LI3E), SupMTI, 10090 Rabat, Morocco. Euro-Mediterranean University of Fes, 30070 Fez, Morocco.
Khalil Azzaoui Laboratory of Engineering Electrochemistry, Modeling and Environment, Faculty of Science, Sidi Mohamed Ben Abdellah University, 30000, Fez, Morocco.
Shehdeh Jodeh Department of Chemistry, An-Najah National University, Nablus, Palestine.
|
*E-mail: [email protected]
Abstract
The objective of this study is to identify potential targets within the SARS-CoV-2 RdRp for the discovery of novel inhibitors derived from therapeutic natural compounds sourced from aromatic and medicinal plants. In the course of this study, a library was generated containing five naturally occurring flavone derivatives (4a, 4b, 4c, 4d, and 4e) in conjunction with the standard favipiravir-RTP. The compounds underwent an in-silico methodology involving critical steps, including (a) evaluating ADME parameters and ensuring compliance with Lipinski’s rule of five, (b) conducting molecular docking analysis, and (c) performing a molecular dynamics simulation lasting 100 nanoseconds. The results suggest that the top five compounds displayed a more favorable pharmacological response compared to the standard, presenting promising outcomes without identified limitations. Consequently, two flavone derivatives (4d and 4e) were chosen due to their higher binding energies compared to the reference molecule, exhibiting binding affinities of -7.036 kcal/mol and -7.141kcal/mol, respectively. Subsequently, the stability of these leading compounds bound with SARS-CoV-2 RdRp was validated through molecular dynamics (MD) simulations, revealing a consistent trajectory (RMSD, RMSF) and favorable molecular properties in their interaction profiles. The present study has identified certain compounds sourced from aromatic and medicinal plants that demonstrate in-silico potential against SARS CoV-2 RdRp. These findings indicate their appropriateness for subsequent in vitro and in vivo evaluations as potential candidates for the treatment of COVID-19 patients.
Keywords: SARS-CoV-2 RdRp, Flavone derivatives, Favipiravir- RTP, ADME analysis, Molecular docking, Molecular dynamics simulation
Introduction
The Covid-19 pandemic is caused by the SARS-CoV-2 virus, which relies on the RNA-dependent RNA polymerase (RdRp) protein for replication (Vicenti et al., 2021). Inhibiting RdRp activity has garnered interest as a potential therapeutic strategy for COVID-19, leading to the development of various nucleoside analog inhibitors authorized for emergency use. Structural studies of RdRp have provided insights into its mechanism of action and potential drug-binding sites (Eastman et al., 2020). However, challenges such as drug resistance development and potential toxicity persist. Favipiravir, a broad-spectrum antiviral medication, has shown promising activity against the SARS-CoV-2 RdRp protein, it undergoes a metabolic transformation into favipiravir-RTP, an active form that serves as a nucleoside analog inhibitor of RdRp activity (Peng et al., 2021). The incorporation of favipiravir-RTP into the growing RNA chain results in the early termination of RNA synthesis and the inhibition of viral replication (Goswami, 2021). Clinical trials have demonstrated favorable outcomes in reducing symptom duration and promoting viral clearance in COVID-19 patients. In vitro studies have indicated that favipiravir-RTP can inhibit SARS-CoV-2 replication (Naydenova et al., 2021). Therefore, this research, approved by the US Food and Drug Administration (FDA), aims to assess the binding capacity of a group of five flavone derivatives compared to the standard Favipiravir-RTP (control) (Hashemian et al., 2021) 7. Analyzing their ability to interact with and inhibit SARS-CoV-2 RdRp targets is crucial for further understanding their potential effectiveness. Favipiravir-RTP has the potential to be a successful COVID-19 treatment, but its efficacy and safety are still under investigation (Merzouki et al., 2023). Moreover, the possibility of the development of drug resistance remains a concern (Kumar et al., 2020). Nonetheless, the history of RdRp inhibition highlights the importance of rapid scientific collaboration and drug development in responding to a global health crisis. Ongoing drug development efforts and continued research into the mechanisms of action of RdRp inhibitors may ultimately lead to effective treatments for this devastating disease. Flavonoids are a category of herbal compounds located in lots of plants, fruits, and vegetables which have demonstrated various health benefits (Adamson et al., 2021), including antiviral activity (Havsteen, 2002). Recent studies have shown that flavone derivatives, such as that extracted from Artemisia herba alba asso (Kumar & Pandey, 2013), demonstrate effective inhibitory properties against COVID-19's causal agent, SARS-CoV-2. Furthermore, flavones impede the activity of the crucial viral RNA-dependent RNA polymerase, a pivotal factor in viral replication, thereby hindering the reproduction of viruses (Merzouki et al., 2023). Additionally, flavone derivatives exhibit immunomodulatory and anti-inflammatory effects, providing relief for COVID-19 symptoms (Faisal et al., 2022). Leveraging flavone derivatives in the treatment of COVID-19 represents a promising therapeutic approach due to their broad-spectrum antiviral activity and favorable safety profile. Flavones are naturally present in various human food sources (Pisoschi et al., 2022), and their regular consumption is associated with numerous health benefits. Moreover, their anti-inflammatory effects contribute to mitigating the body's inflammatory response in various chronic conditions. In the context of drug development, in silico techniques have become powerful tools, especially in the battle against SARS-CoV-2. Given the essential role of the RdRp protein in viral replication, it serves as a potent target for antiviral medications. Ensuring the effectiveness and safety of a drug requires specific ADME (Absorption, Distribution, Metabolism, and Excretion) properties (Yao et al., 2004). In the early stages of drug development, in silico technologies play a crucial role in predicting these properties. Molecular docking, another in silico technique, helps identify potential drug candidates that bind to the RdRp protein (Wang et al., 2015). By simulating interactions between a molecule and the target protein, researchers can pinpoint compounds with the potential to inhibit viral replication. Molecular dynamics simulations further contribute to the understanding of RdRp protein behavior and its interactions with potential drug candidates (Qayed et al., 2022). These simulations shed light on the dynamics and stability of the protein-ligand complex, as well as the putative drug's mechanism of action (Mosquera-Yuqui et al., 2022). Numerous recent in silico research projects have been conducted to identify suitable RdRp inhibitors for treating SARS-CoV-2. Some of these investigations have led to the discovery of promising medication candidates, several of which are currently undergoing clinical trials.
Materials and Methods
Ligand Preparations and Protein Preparations
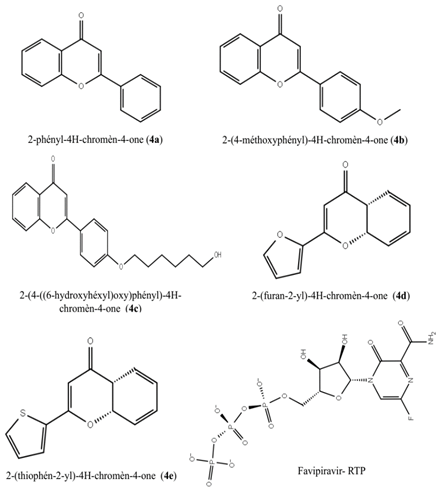
In summary, all five ligands of natural flavone derivatives and the standard favipiravir-RTP obtained from PubChem (CID: 5271809) (Figure 1) were acquired in SDF format and imported into Maestro 12.8(Schrodinger2021-2) for ligand preparation in the docking process (Gul et al., 2021). The ligand production not only facilitates the generation of low-energy structures but also allows for the extension of each input structure to meet required stereochemistry by producing tautomers and confirming changes in ring ionization states. To establish ionization states for each ligand structure at physiological pH 7±2 units, Schrödinger Maestro's LigPrep wizard (Schrodinger2021-2) was employed, utilizing Epic. The ligands underwent minimization using the OPLS2005 force field, with other options left at their default settings. For the subsequent docking with Glide, output files generated from the ligand minimization stage were utilized. The SARS-CoV-2 RdRp protein (ID: 7AAP) (Figure 2), depicting the Nsp7-Nsp8-Nsp12 SARS-CoV-2 RNA-dependent RNA polymerase in conjunction with template: primer dsRNA and favipiravir-RTP, was obtained from the PDB (Protein Data Bank) with a crystalline structure resolution of 2.50 Å (Diass et al., 2023) 23. The protein is comprised of four chains, with the catalytic domain situated in chain A (nsp12). Chain B and Chain D represent nsp8, while chain C represents nsp7. Upon importing into Schrödinger Maestro for docking tests, the proteins underwent pretreatment, which included the initial assignment of hydrogen bonds. Protein optimization ensued through the assignment of hydrogen bonds using PROPKA at pH=7.0. Subsequently, protein analysis was conducted through restricted minimization utilizing the OPLS-2005 methodology (Parise et al., 2022).
|
|
|
Figure 1. The molecular configurations of the flavone derivatives employed in the present investigation and the standard compound. |
Determining ADME Properties
The QikProp module within the Schrödinger 2021-2 software was employed to assess the ADME (Absorption, Digestion, Metabolism, Excretion) features (Basha et al., 2014), comparing the top five compounds with the standard favipiravir-RTP. The software's input zone sketcher allows users to create, modify, and import 2-dimension structures, and the resulting output files can be saved in the Structure Data File (SDF) format, Haut du formulaire Compliant with various visualization applications. This study emphasizes crucial physicochemical properties such as Flexibility, Molecular Weight, Bioavailability, Hydrophobicity, Permeability, and Polar Solubility.
The compounds that underwent the most extensive study were scrutinized for violations of Lipinski's Rule of Five to underscore their potential (Fajriyah et al., 2023).
|
|
|
Figure 2. 3D structure of SARS-CoV-2 RdRp (7AAP) with the active site employed for docking molecular in the present study |
Molecular Docking Calculation
For the docking process, the protein, which had been previously reduced and optimized in the preceding stage, was employed. The initial step towards docking involved preparing a grid with the exact coordinates matching the native location of 7AAP (Favipiravir-RTP). In the central position of the grid box, sized 10 Å × 10 Å × 10 Å, the orientation was established with the coordinates provided: 108.14° on the X-axis, 103.27° on the Y-axis, and 105.87° on the Z-axis. Grids for docking, integrating the specified parameters, were generated using the "receptor grid generation" feature within the Glide module in Schrödinger Maestro (Faris et al., 2023). Following this, the 'ligand docking' window of the Glide module in Schrödinger Maestro was filled with the output files from the receptor grid generation, along with the ligands that had been minimized and prepared for docking. The settings included an adjustable ligand sampling, an epic state penalty for the docking score, and the standard precision protocol (SP) for docking precision. To visualize only the best poses, the output was refined. The glide module in Schrödinger was employed for all docking procedures, the optimal protein-ligand docking schematics were presented in both 2D and 3D, visualized in BIOVIA Discovery Studio, 2021, showcasing interacting complexes sorted by assembly analysis (Cherriet et al., 2023).
Molecular Dynamics Study of the Best Docked Complex
A molecular dynamics (MD) simulation lasting 100 nanoseconds was performed to validate the stability of the best-docked conformations of the drug across all six sets of docked results. This included both the docked complexes and the standard favipiravir-RTP with the receptor. The objective was to gain insights into the interaction and stability of the protein-ligand complex. The MD simulation was carried out using the system builder tool and the Desmond module from the Schrödinger 2021-2 suite, operating on a Linux operating system (Boumezzourh et al., 2023). The OPLS_2005 force field was utilized to simulate the TIP3P (transferable intermolecular interaction potential 3 points) solvent model within an orthorhombic box with dimensions of 10 Å × 10 Å × 10 Å. Counter ions were added as needed to neutralize the models, and 0.15 M sodium chloride was introduced to replicate physiological conditions. The Smooth Particle Mesh Ewald (PME) approach was employed with a temperature of 300 K and a pressure of 1.01325 bar for the simulation. The simulations were conducted under isothermal-isobaric ensemble NPT (constant number of particles, pressure, and temperature) conditions. Specific equations were applied to calculate the root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) trajectories, providing insights into the dynamics and stability of the protein-ligand interaction.
|
|
(1) |
|
|
(2) |
Following the simulation, plots, and figures depicting the interaction profiles between the ligands and proteins, as well as visualizations of RMSD, RMSF, and the count of hydrogen bonds, were generated utilizing the Simulation Interaction Diagram wizard.
Results and Discussion
In Silico Predictions ADME of Pharmacokinetic Parameters
Due to its efficiency and cost-effectiveness, the utilization of ADME modeling has become increasingly prevalent in pharmaceutical research for the development of drugs. Table 1 present the outcomes of Lipinski's rule of five and the ADME predictions from Qikprop for both the five flavone derivatives and the standard favipiravir-RTP. Parameters such as molecular weight, hydrogen bond donors, hydrogen bond donor acceptors, and the expected octanol/water partition coefficient were all projected to precisely adhere to Lipinski's rule of five., showing no violations in the analysis of the five flavones. However, the control, favipiravir-RTP, deviated from Lipinski's rule with predicted values exceeding the threshold for molecular weight (529.159 > 500), hydrogen bond acceptors (21.6 > 10), and QPlogPo/w (28.061 > 5), indicating rule violations for these parameters in the control compound. All 5 flavones with values were discovered in a range that complies with the requirements as stated In the investigation of surface components outlined in Schrödinger's Qikprop manual, which includes parameters such as SASA (solvent-accessible surface area), FOSA (fractional molecular surface area), FISA (fractional internal surface area), PISA (polar internal surface area), PSA (polar surface area), and volume, it was observed that predicted qualitative human oral absorption was high for all five flavones. The QPPCaco values (predicted apparent Caco-2 cell permeability) were greater than 500 nm/sec, indicating excellent permeability, and the QPPCaco (gut barrier model) value was found in an acceptable range between -3.0 and +1.2.
Standard favipiravir-RTP showed poor permeability prediction values with respect to QPPCaco and QPlogBB, and the negative values indicate that the compound is polar and has low permeability, low predicted qualitative human oral absorption (HOA). In addition, FOSA and PSA values were not found within an acceptable range see Table 2.
Regarding the Volume (Total solvent-accessible volume) all 5 flavones and the favipiravir-RTP have values in the favorable range (500– 2000).
Overall, in contrast to favipiravir-RTP, the five flavone derivatives demonstrated "good" Lipinski rule of five values and predicted pharmacokinetic parameters without any violations with drug-like characteristics as infection inhibitors against SARS-CoV-2. This study suggests that potential in vitro compounds may be further used for preclinical evaluation.
Table 1. In silico Lipinski's rule of five and Predicted ADME analysis for potential compounds.
|
Compound Name |
(4a) |
(4b) |
(4c) |
(4d) |
(4e) |
Favipiravir- RTP |
|
Molecular Weiǥht |
222.243 |
252.269 |
338.402 |
214.220 |
230.281 |
529.159 |
|
Donor HB |
0 |
0 |
1 |
0 |
0 |
3 |
|
Acceptor HB |
2.50 |
3.25 |
4.95 |
3.25 |
2.75 |
21.6 |
|
QPlogPo/w |
6.407 |
6.640 |
8.951 |
6.553 |
6.044 |
28.061 |
|
Rule of five |
0 |
0 |
0 |
0 |
0 |
3 |
|
SASA |
458.8 |
496.2 |
667.4 |
424.0 |
452.7 |
716.2 |
|
FOSA |
00.00 |
92.58 |
219.0 |
40.01 |
38.80 |
70.53 |
|
FISA |
54.88 |
54.82 |
111.1 |
50.88 |
50.39 |
569.2 |
|
PISA |
403.9 |
348.4 |
337.2 |
333.1 |
318.7 |
213.1 |
|
PSA |
35.46 |
43.88 |
66.21 |
44.04 |
35.32 |
313.7 |
|
Volume |
757.358 |
832.444 |
1150.48 |
698.597 |
744.481 |
1263.32 |
|
QPPCaco |
2988.512 |
2992.438 |
875.4010 |
3260.850 |
3296.199 |
0.001000 |
|
QPlogBB |
0.086 |
0.012 |
-1.089 |
0.143 |
0.237 |
-6.241 |
Molecular Dockinǥ Results Analysis
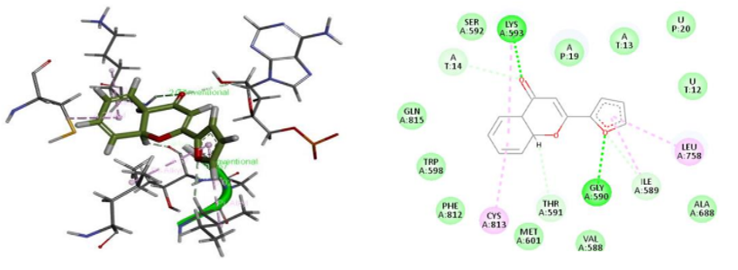
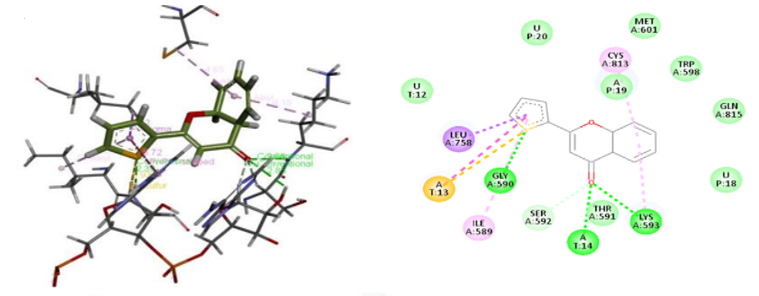
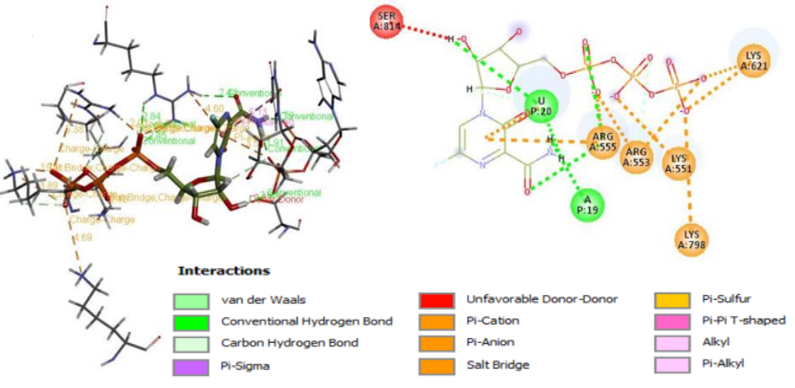
A strategy that is frequently utilized to comprehend the interactions between ligands and receptors during the drug discovery process is the molecular docking in silico technique. In the pursuit of drug discovery for emerging and severe diseases such as SARS-CoV-2, techniques like virtual screening and drug repurposing are actively employed. Moreover, drug repurposing proves to be an efficient strategy in the quest for new drugs due to its cost-effectiveness and accelerated process. This study focused on identifying potential drugs capable of inhibiting SARS-CoV-2 RdRp through the application of molecular docking techniques. The evaluation of ligand-receptor interaction strength involves the utilization of various scores, including docking score and glide model, facilitated by the Glide module within Schrödinger's docking research framework. The docking score, representing a scoring function, is instrumental in predicting the binding affinity between targets and ligands. A more negative docking score correlates with a higher binding affinity. The glide model, emphasizing weighted Van der Waals and electrostatic energies, is particularly valuable for comparing conformers but less so for contrasting chemically distinct species. Given this consideration, Glide employs Emodel to select the optimal poses for the ligand, while DockScore is employed to compare these chosen poses against each other. Using glide mode with Standard Precision (SP), molecular docking studies were carried out on the five flavone derivatives and authorized standard favipiravir-RTP from the PubChem database on the SARS-CoV-2 RdRp enzyme binding pocket (PDB ID: 7AAP) to identify suitable candidates for the treatment of COVID-19. Comparing the outcomes to favipiravir-RTP. The outcomes of the docking analysis are displayed in Table 2. The amino acids of the SARS-CoV-2 RdRp were extremely stable inside the cavity, and the docking study revealed that all five flavone derivatives were docked at the profile active site. The most stable compounds are 4d and 4e which have docking score values of -7.036kcal/mol and -7.141kcal/mol, respectively, greater than the docking score value of -6.068 kcal/mol for the control compound, favipiravir-RTP (Figure 3). We begin with the flavone derivative 4e. Oxygen and sulfur interact with LYS593 and GLY590 by conventional hydrogen bonds, respectively. A carbon-hydrogen bond is formed with SER592, two interactions are Alkyl with CYS813 and LYS593, and two more interactions are Pi-Alkyl and Pi-Sigma with the following residues ILE580 and LEU758. the following residues THR561 and MET601 were mainly involved in the interaction with Van Der Waals. For the 4d formed two Conventional Hydrogen bonds with LYS593 and GLY590, and two Carbon Hydrogen bonds with TRH591 and ILE589, two interaction alkyl with CYS813 and LYS593, and two other Pi-Alkyl interactions with the following residues ILE580 and LEU758, residues SER592, AL688, VAL588, MET601, PHE812, TRP598 and GLN815 are involved in the interaction Van Der Waals. For the remaining three compounds 4a, 4b, and 4c are stable in the active site but the binding energy is lower than that of the standard -4.663kcal/mol, -4.399kcal/mol and -4.358kcal/mol respectively, and their interactions with various amino acid residues are shown in Table 2. To ascertain the therapeutic efficacy of these flavone derivative drugs against SARS CoV-2 RdRp, more clinical and in vivo research is necessary.
Table 2. The binding affinities and the contributing binding residues of SARS CoV-2 RdRp with selected compounds were generated using SP docking.
|
Compound Name |
Dock Score (Kcal/mol) |
Glide emodel |
Contributing Binding Residues |
H-bonds |
Bond Distance (Å) |
|
(4a) |
-4.663 |
-30.240 |
ARG346, PRO461, PRO323, PRO677, ASN628 GLU350, ASN314, VAL315, SER318, PHE326, YHR319, VAL320, PHE321, PRO322. |
0 |
0 |
|
(4b) |
-4.399 |
-37.489 |
SER397, TYR149, PHE396, LYS391, PHE396, CYS395, LEU389, THR393, ASP390, THR394, THR137. |
SER397 |
2.21 |
|
(4c) |
-4.358 |
-49.539 |
ILE266, PRO323, VAL315, PRO461, ARG349, PRO461, SER255, TRP268, SER318. |
ILE266 |
2.02 |
|
(4d) |
-7.036 |
-50.242 |
LYS593, GLY590, THR591, ILE589, CYS813, SER592, ALA688, VAL588, MET601, PHE812, TRP598, GLN815. |
LYS593 GLY590 |
2.77 2.67 |
|
(4e) |
-7.141 |
-54.781 |
LYS593, GLY590, LEU758, CYS813, ILE580, SER592, YHR581, MET601, LYS593. |
LYS593 GLY590 |
2.68 2.39 |
|
Favipiravir- RTP |
-6.068 |
-101.04 |
ARG555, ARG553, LYS798, LYS813, LYS621, CYS813, LYS545, ARG836, PRO620, SER549, SER814. |
ARG555 ARG555 |
2.84 2.45 |
|
|
|
a) |
|
|
|
b) |
|
|
|
c) |
|
Figure 3. 2D and 3D interactions of ligands (a) 4d, (b) 4e, and (c) standard with the receptor protein. |
Molecular Dynamics Simulation Assessment
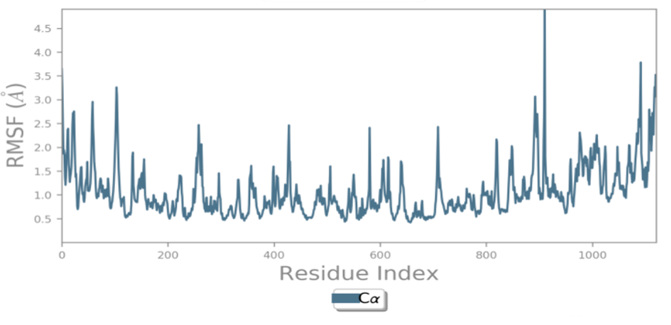
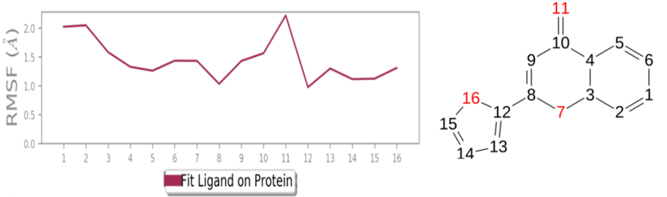
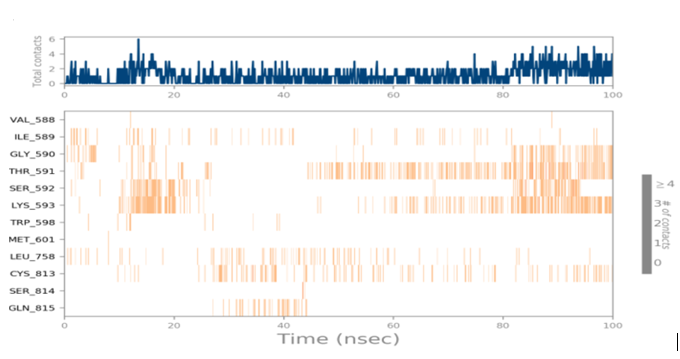
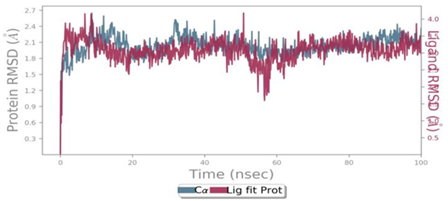
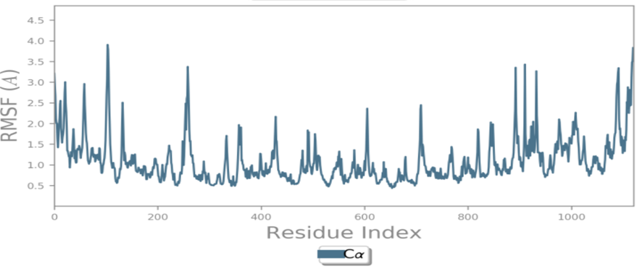
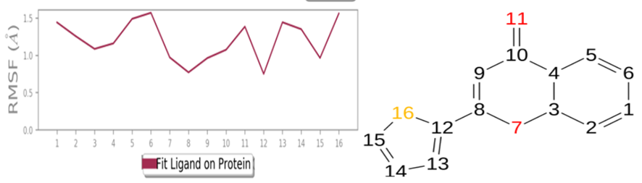
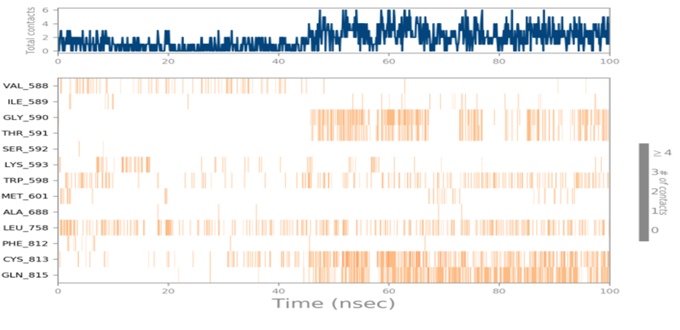
MD simulations are utilized to rigorously evaluate whether the proposed certainty by the docking assessment for ligand-protein interaction manifests in reality or not. By using these simulations, we can be certain that the expected interaction length and type correspond to the docking results. By determining the robustness of the interaction, MD simulations help to overcome the constraint that docking has in that it can only anticipate the types of interactions that might occur between a protein and ligand. Additionally, MD simulations can be used to examine the stability over time of the ideal docking orientation of the interacting protein and the ligand. The study measured the RMSD of the protein (left Y-axis) and the ligand (right Y-axis) in order to examine molecular dynamics simulations. Together with the RdRp SARS CoV-2 with 4d and the 4e (flavone derivatives) greatest docking findings were achieved, as well as the RMSF and the protein ligand interactions as a function of time. The structural variation over a while of 0 to 100 ns was measured using the RMSD values of the complexes. The protein-ligand complexes remained stable and in touch with one another throughout the simulation period, with no appreciable fluctuations being noticed. The shape of a protein affects its stability, as seen in Figures 4a and 5a. Larger proteins can exhibit a wide range of RMSD values, indicating a well-balanced nature of the protein. Changes in RMSD values of the protein backbone within the range of 1-4 are common for small lobular proteins. To find significant changes in the protein domain, the RMSF was used. Except for the terminal amino acid residues (the tails N- and C), the SARS-CoV-2 RdRp displayed an RMSF value ranging from 0.5 to 2.5. Usually, secondary structural components such as alpha helices and beta strands are more rigid compared to the unstructured regions of the protein. Consequently, these components undergo fewer changes than loop regions. Which were observed to move faster than other parts of the protein. This likely contributed to the observed variation in RMSF values for these residues. Despite this, the protein was generally found to exhibit good flexibility over the trajectory. In the present investigation, the “Lig fit Prot” values of 4d and 4e are good and acceptable because the values do not exceed 1.5 Å and 2 Å, respectively. Thus, from this, it can be concluded that the two flavone derivatives interact decisively with a docking score. Finally, as seen in (Figure 4) and (Figure 5), a timeline of complex interactions is shown during the simulation period of 100 ns in the form of Hydrogen bonding, Hydrophobic contact, Ionic interactions, and Water bridges contribute to the formation of distinct contacts between the protein and the ligand during the trajectory, as depicted in the upper panel. Additionally, multiple residues created numerous unique interactions with the two flavone derivatives, which are depicted by a dark orang-like color. There are other forms of connections as well, and the amino acids GLY590, THR561, LEU758, CYS813, and GLN815 formed the hydrogen bond in less than 40 ns. In this instance, only GLY590 and LYS593 were projected to interact strongly based on the docking evaluation but interacted very moderately during the simulation. Instead, strong connections developed during molecular dynamic simulations but weren't anticipated by docking. RdRp interacted successfully with 4d and 4e overall. SARS-CoV-2. Due to the requirement for additional safety precautions and ethical permission, working with SARS-CoV-2 in a laboratory setting presents practical challenges. In such a case, a complex in silico workflow involving assessments of the ADME properties, docking molecules, and MD simulations is assisting us in making highly accurate predictions about the behavior of the SARS-CoV-2 drugs.
|
|
|
a) |
|
|
|
b) |
|
|
|
c) |
|
|
|
d) |
|
Figure 4. Through the application of Molecular Dynamics (MD) studies, the stability of the docked complex of 4d - RdRp was investigated, focusing on (a) Protein-ligand RMSD, (b) Protein RMSF, (c) Ligand RMSF, and (d) Protein-ligand contacts. |
|
|
|
a) |
|
|
|
b) |
|
|
|
c) |
|
|
|
d) |
|
Figure 5. Through the application of Molecular Dynamics (MD) studies, the stability of the docked complex of 4e - RdRp was investigated, focusing on (a) Protein-ligand RMSD, (b) Protein RMSF, (c) Ligand RMSF, and (d) Protein-ligand contacts. |
Conclusion
Screening for potential drugs targeting COVID-19, a problem on a global scale for the prediction of drug binding to a particular or single target of RNA viruses, is the subject of numerous initiatives and research projects. Since favipiravir-RTP's usual target is SARS-CoV-2 RdRp, the current study's rigorous computational methodology sought to anticipate the bindin' potential of five ligands of natural and synthetic flavone derivatives towards that target. Five potential compounds (4a, 4b, 4c, 4d, and 4e) were selected from which two potential inhibitors of SARS-CoV-2 RdRp showed commendable compared with favipiravir- RTP, docking scores, these are Dock Score(4d) = -7.036 kcal/mol and Dock Score(4e) = -7.141 kcal/mol, which exhibits good binding affinity towards the active site. The five flavone derivatives were subsequently examined using various methodologies employed, including Lipinski's rule of five and ADME parameters, to identify potential drugs with a range of pharmacological features. Furthermore, during MD simulations, the study of RMSD, RMSF, and hydrogen bond wells revealed that two flavone derivatives were in a more stable interaction with SARS-CoV-2 RdRp. All these properties strengthen our motivation to use flavones as potential drugs against COVID-19, and we truly believe that this in silico research will lead to the development of therapeutics for COVID-19.
Acknowledgments: None
Conflict of interest: None
Financial support: None
Ethics statement: None
Adamson, C. S., Chibale, K., Goss, R. J., Jaspars, M., Newman, D. J., & Dorrington, R. A. (2021). Antiviral drug discovery: Preparing for the next pandemic. Chemical Society Reviews, 50(6), 3647-3655.
Basha, S. H., Bethapudi, P., Majji Rambabu, F. (2014). Anti-angiogenesis property by Quercetin compound targeting VEGFR2 elucidated in a computational approach. European Journal of Biotechnology and Bioscience, 2, 30-46.
Boumezzourh, A., Ouknin, M., Merzouki, M., Dabbous-Wach, A., Hammouti, B., Umoren, P. S., Costa, J., Challioui, A., Umoren, S. A., & Majidi, L. (2023). Acetylcholinesterase, tyrosinase, α-Glucosidase inhibition by ammodaucus leucotrichus coss. & dur. fruits essential oil and ethanolic extract and molecular docking analysis. Moroccan Journal of Chemistry, 11(04), 897-1318.
Cherriet, S., Merzouki, M., El-Fechtali, M., Loukili, E., Challioui, A., Soulaymani, A., Nanadiyanto, A. B. D., Ibriz, M., Elbekkaye, K., & Ouasghir, A. (2023). In Silico Investigation of Aristolochia longa Anticancer Potential against the Epidermal Growth Factor Receptor (EGFR) in the Tyrosine Kinase Domain. Moroccan Journal of Chemistry, 11(04), 1074-1085.
Diass, K., Merzouki, M., Elfazazi, K., Azzouzi, H., Challioui, A., Azzaoui, K., Hammouti, B., Touzani, R., Depeint, F., Ayerdi Gotor, A., et al. (2023). Essential oil of lavandula officinalis: Chemical composition and antibacterial activities. Plants, 12(7), 1571. doi:10.3390/plants12071571
Eastman, R. T., Roth, J. S., Brimacombe, K. R., Simeonov, A., Shen, M., Patnaik, S., & Hall, M. D. (2020). Remdesivir: A review of its discovery and development leading to emergency use authorization for treatment of COVID-19. ACS Central Science, 6(5), 672-683.
Faisal, S., Badshah, S. L., Kubra, B., Sharaf, M., Emwas, A. H., Jaremko, M., & Abdalla, M. (2022). Computational study of SARS-CoV-2 rna dependent rna polymerase allosteric site inhibition. Molecules, 27(1), 223.
Fajriyah, N. N., Mugiyanto, E., Rahmasari, K. S., Nur, A. V., Najihah, V. H., Wihadi, M. N., Merzouki, M., Challioui, A., & Vo, T. H. (2023). Indonesia herbal medicine and its active compounds for anti-diabetic treatment: A systematic mini review. Moroccan Journal of Chemistry, 11(04), 948-964.
Faris, A., Edder, Y., Louchachha, I., Lahcen, I. A., Azzaoui, K., Hammouti, B., Merzouki, M., Challioui, A., Boualy, B., Karim, A., et al. (2023). From himachalenes to trans-himachalol: unveiling bioactivity through hemisynthesis and molecular docking analysis. Scientific Reports, 13(1), 17653.
Goswami, D. (2021). Comparative assessment of RNA-dependent RNA polymerase (RdRp) inhibitors under clinical trials to control SARS-CoV2 using rigorous computational workflow. RSC Advances, 11(46), 29015-29028.
Gul, S., Ozcan, O., Asar, S., Okyar, A., Barıs, I., & Kavakli, I. H. (2021). In silico identification of widely used and well-tolerated drugs as potential SARS-CoV-2 3C-like protease and viral RNA-dependent RNA polymerase inhibitors for direct use in clinical trials. Journal of Biomolecular Structure and Dynamics, 39(17), 6772-6791.
Hashemian, S. M., Farhadi, T., & Velayati, A. A. (2021). A review on favipiravir: the properties, function, and usefulness to treat COVID-19. Expert Review of Anti-Infective Therapy, 19(8), 1029-1037.
Havsteen, B. H. (2002). The biochemistry and medical significance of the flavonoids. Pharmacology & Therapeutics, 96(2-3), 67-202.
Kumar, B. K., Sekhar, K. V. G. C., Kunjiappan, S., Jamalis, J., Balaña-Fouce, R., Tekwani, B. L., & Sankaranarayanan, M. (2020). Druggable targets of SARS-CoV-2 and treatment opportunities for COVID-19. Bioorganic Chemistry, 104, 104269.
Kumar, S., & Pandey, A. K. (2013). Chemistry and biological activities of flavonoids: An overview. The Scientific World Journal, 2013.
Merzouki, M., Challioui, A., Bourassi, L., Abidi, R., Bouammli, B., & El Farh, L. (2023). In silico evaluation of antiviral activity of flavone derivatives and commercial drugs against SARS-CoV-2 main protease (3CLpro). Moroccan Journal of Chemistry, 11(1), 129-143.
Mosquera-Yuqui, F., Lopez-Guerra, N., & Moncayo-Palacio, E. A. (2022). Targeting the 3CLpro and RdRp of SARS-CoV-2 with phytochemicals from medicinal plants of the Andean Region: molecular docking and molecular dynamics simulations. Journal of Biomolecular Structure and Dynamics, 40(5), 2010-2023.
Naydenova, K., Muir, K. W., Wu, L. F., Zhang, Z., Coscia, F., Peet, M. J., Castro-Hartmann, P., Qian, P., Sader, K., Dent, K., et al. (2021). Structure of the SARS-CoV-2 RNA-dependent RNA polymerase in the presence of favipiravir-RTP. Proceedings of the National Academy of Sciences, 118(7), e2021946118.
Parise, A., Ciardullo, G., Prejanò, M., Lande, A. D. L., & Marino, T. (2022). On the recognition of natural substrate CTP and endogenous inhibitor ddhCTP of SARS-CoV-2 RNA-dependent RNA polymerase: A molecular dynamics study. Journal of Chemical Information and Modeling, 62(20), 4916-4927.
Peng, Q., Peng, R., Yuan, B., Wang, M., Zhao, J., Fu, L., Qi, J., & Shi, Y. (2021). Structural basis of SARS-CoV-2 polymerase inhibition by favipiravir. The Innovation, 2(1), 100080.
Pisoschi, A. M., Pop, A., Iordache, F., Stanca, L., Geicu, O. I., Bilteanu, L., & Serban, A. I. (2022). Antioxidant, anti-inflammatory and immunomodulatory roles of vitamins in COVID-19 therapy. European Journal of Medicinal Chemistry, 232, 114175.
Qayed, W. S., Ferreira, R. S., & Silva, J. R. A. (2022). In silico study towards repositioning of FDA-approved drug candidates for anticoronaviral therapy: Molecular docking, molecular dynamics and binding free energy calculations. Molecules, 27(18), 5988.
Vicenti, I., Zazzi, M., & Saladini, F. (2021). SARS-CoV-2 RNA-dependent RNA polymerase as a therapeutic target for COVID-19. Expert Opinion on Therapeutic Patents, 31(4), 325-337.
Wang, Y., Xing, J., Xu, Y., Zhou, N., Peng, J., Xiong, Z., Liu, X., Luo, X., Luo, C., Chen, K., et al. (2015). In silico ADME/T modelling for rational drug design. Quarterly Reviews of Biophysics, 48(4), 488-515.
Yao, L. H., Jiang, Y. M., Shi, J., Tomas-Barberan, F. A., Datta, N., Singanusong, R., & Chen, S. S. (2004). Flavonoids in food and their health benefits. Plant Foods for Human Nutrition, 59, 113-122.