In-silico Prediction of Drug Target, Molecular Modeling, and Docking Study of Potential Inhibitors against Burkholderia pseudomallei
|
Raghunath Satpathy* School of Biotechnology, Gangadhar Meher University, Amruta Vihar, Sambalpur Odisha, 768004, India. *E-mail: [email protected] |
Abstract
The infection of the Burkholderia pseudomallei causes the disease melioidosis. for which the treatment method takes longer time, and sometimes it is difficult to completely eradicate the bacteria from the body. Moreover, its antibiotic resistance in nature created great concern in recent times. Hence, there is an urgent requirement to identify new drug molecules that can improve the current process of treatments and reduce the risk to people. This study analyzed the pyrimidine metabolic pathways of Burkholderia pseudomallei strain K96243, and UMP (Uridine monophosphate/Uridylate) kinase enzyme was selected as the drug target. After structure prediction by the AlphaFold server, the validation of the structure was performed by using Procheck, Verify3D, and Errat tool. Further, six probable inhibitor molecules were selected from the PubChem database, including the natural inhibitor of the enzyme, Uridine triphosphate (UTP). The molecular docking study predicted that the UTP (CID 6133) had the highest docking score, followed by another compound PubChem (Compound ID) CID 284262. Then, Toxicity and ADMET properties were computed and analyzed. Further, a 5 nanosecond molecular dynamics simulation of the complex of UMP-Kinase and CID 284262 was performed by using the Gromacs 5.1.1 software to analyze the stability of the best complex. It was predicted that the CID 284262 might be considered a suitable inhibitor of the enzyme.
Keywords: Metabolic pathway analysis, UMP kinase enzyme, Molecular docking, Drug target prediction, Molecular dynamics simulation, Inhibitor compounds
Introduction
Burkholderia pseudomallei is a pathogenic gram-negative, bi-polar, aerobic, motile, rod-shaped soil-dwelling bacterium, and the infection in the tropical and subtropical regions causes the disease melioidosis (Wiersinga et al., 2018; Savelkoel et al., 2022). These bacteria are also known for the resistant to various environmental conditions, including deficiency of nutrients, extreme temperature, and pH (Inglis et al., 2001). It infects humans and animals, primarily cattle and livestock such as goats, pigs, and sheep (Ekakoro et al., 2022). Bacterial infection in humans is associated with different pathogenic conditions like widespread pulmonary infection, septicemic infection, diabetic patients, and renal disease (Gassiep et al., 2020). Continuous exposure to infected soils and groundwater leads to an increased risk of causing melioidosis. Pneumonia is the most common symptom of melioidosis, observed in half of all cases (Leung et al., 2023). The seriousness of the disease varies widely, such as illness with high fever, tiny cough, inflamed pain, and difficulty breathing (Khattab et al., 2022). Presently the drug doxycycline is widely used as one of the most efficient therapeutic strategies for infection (Sridharan et al., 2021). However, studies reported that the B. pseudomallei bacteria also shows resistance to doxycycline while testing against the drug with different strains (Zueter et al., 2022). This bacterium is also naturally resistant to many antibiotics, including penicillin, cephalosporins, macrolides, rifamycins, polymyxins, and aminoglycosides (Di Fiore et al., 2022). The process of antimicrobial resistance has been studied as the bacterial antibiotic efflux pump was characterized as the key cause of the resistance (Somprasong et al., 2021). Since no licensed versions of vaccines are available for melioidosis disease, new therapeutic measures and vaccination strategies are expected to come shortly as a preventive measure against the infection (Morici et al., 2019; Currie 2022). In drug design strategies against bacteria, nucleotide metabolism is frequently studied to identify the drug targets, as the pathway's enzymes are considered essential (Kumari, & Tripathi, 2021; Perveen & Sharma, 2022). The nucleoside monophosphate (NMP) kinases are a critical group of enzymes involved in the pyrimidine synthesis metabolic process (Beji et al., 2020). The de novo of the pyrimidine nucleotide biosynthetic process consists of different vital metabolic enzymes conserved among the prokaryotic and eukaryotic organisms, including humans (Uddin et al., 2020; Wyllie et al., 2022).
The current work aims to discover some inhibitor molecules from the database and investigate the inhibition potential against the UMP kinase of Burkholderia pseudomallei (strain K96243) bacteria.
Materials and Methods
Computational Resources Used for Molecular Docking and MD Simulation Study
Processor: AMD Ryzen 3900*4.6GHz, Mother Board: Gigabyte B550 Acurs pro AC, RAM: Corsair 16*2 vengeance 3200NH 32GB, GPU: Asus dual GT 165004G, Operating system: Ubuntu Version 2021
Analysis of Nucleotide Synthesis Metabolic Pathway of Burkholderia Pseudomallei (strain K96243) and Target Prediction
The nucleotide synthetic pathway of the Burkholderia pseudomallei was searched in the KEGG pathway database (https://www.genome.jp/kegg/) with the organism keyword. The active genes involved in the pyrimidine metabolism were retrieved along with the sequence and functional information. Further, the sequence similarity was searched against the Homo sapiens using the protein Basic Local Alignment Search Tool (BLAST) available at https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins by using the default parameter. The essential enzymes of the bacteria having no significant similarity were selected as the target in the present study. Further, the essential nature of the selected target protein was searched in the DEG (Database of Essential gene) database (http://origin.tubic.org/deg/public/index.php) to confirm the result.
Retrieval of the Three-Dimensional (3D) Structure of the Target Enzyme of Burkholderia Pseudomallei (strain K96243) and Validation
The 3D structure of the enzymes was obtained from the AlphaFold server (https://alphafold.ebi.ac.uk/). Further, the structural validation was performed by analyzing the output of several tools like PROCHECK (Ramachandran plot), ERRAT, and Verify 3D (https://saves.mbi.ucla.edu/).
Retrieval of Ligand Information
In the present work, the UTP and five similar types of compounds were considered inhibitor molecules by searching the literature (Arvind et al., 2013). The compound structures were retrieved from the PubChem database, and several molecular features were computed.
Molecular Docking, ADMET Property, and Molecular Dynamics Simulation Study
This selected target protein (receptor) and the ligand were subjected to the molecular docking study. Molecular docking uses searching and scoring algorithms to calculate the binding affinity and conformation of the ligand on the receptor molecule (Satpathy, 2020; Aleidi et al., 2022). Followed by molecular docking, the toxicity level and ADMET (Absorption, distribution, metabolism, excretion, and toxicity) properties were evaluated for the ProTox-II server (https://tox-new.charite.de/protox_II/) and Swiss ADME server (http://www.swissadme.ch/) respectively. Protox II server takes the input molecule in the canonical SMILES (Simplified Molecular-Input Line-Entry System) format. It predicts the compound's toxicity in six classes (Class I fatal to Class VI non-toxic). Further, GROMACS 5.1.1 (Groningen Machine for Advanced Chemical Simulations) software was employed for MD simulation at 5 nanoseconds (ns) time scale. It is an open-source, free molecular dynamics modeling program that is primarily made for simulating biological molecules, including proteins, lipids, and nucleic acids (www.gromacs.org). The 5 nanosecond molecular dynamics simulation was conducted for the receptor only and the selected ligand-receptor complex in the water environment by choosing the GROMOS 43A1 force field with SPCE as the water molecule topology. For the complex simulation, the topology file of the ligand molecule was obtained from the Prodrg server (http://prodrg1.dyndns.org/submit.html). The MD simulation result generated the parameters such as Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), Radius of gyration (Rg), Hydrogen bond, and Solvent accessible surface area (SASA) were considered for further analysis (Daivasigamani et al., 2022; Ghahremanian et al., 2022).
Results and Discussion
Metabolic Pathway Analysis and Identification of the Target
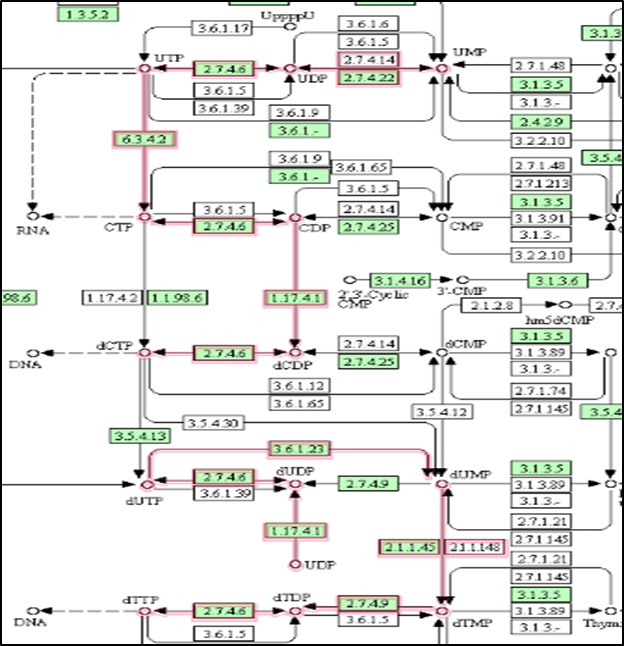
The metabolic pathway related to pyrimidine metabolism resulted in 7 active enzymes involved in the pathways (Figure 1). The enzyme details with the E.C. numbers and functions were retrieved. Further, the protein sequences were obtained from the Uniport database. Further the
|
|
|
Figure 1. Screenshot of the active enzymes (highlighted in red) involved in the pyrimidine metabolism of Burkholderia pseudomallei |
Position-specific iterated Blast (PSI-BLAST) program was used to obtain the homologous nature of the proteins concerning the human genome. The output of the seven selected enzymes is shown in Table 1. The enzyme uridylate kinase (uridine monophosphate kinase) did not show any homology as no significant similarity obtained with the human genome was selected as the target for the present study. The DEG database search also confirms the essential nature of the enzyme as it is available in the database with the DEG ID DEG10350258.
Table 1. Enzyme details involved in the pyrimidine metabolism and selection of targets
|
S. N |
Name of the Enzyme |
Gene |
E.C No |
Function |
BLAST Matching with human, E-value, and % similarity |
|
1 |
nucleoside diphosphate kinase |
ndk |
EC:2.7.4.6 |
Synthesis of nucleoside triphosphates other than ATP. |
Similar to NDP kinase, 9e-34, 43.94% |
|
2 |
Uridylate kinase |
pyrH |
EC:2.7.4.22 |
Catalyzes the reversible phosphorylation of UMP to UDP |
No Significant Similarity |
|
3 |
CTP synthase |
pyrG |
EC:6.3.4.2 |
Catalyzes the ATP-dependent amination of UTP to CTP |
CTP synthase 2, 1e-166, 45.08% |
|
4 |
ribonucleoside-diphosphate reductase beta chain |
nrdB |
EC:1.17.4.1 |
Provides the precursor molecules, essential for DNA synthesis. |
ribonucleoside-diphosphate reductase subunit M2 B isoform 1 1e-38, 26.88% |
|
5 |
dUTP Pyrophosphatase |
dut |
EC:3.6.1.23 |
Generates the immediate precursor, dUMP, of thymidine nucleotides |
dUTP Pyrophosphatase complex with dUDP 1e-17,35.88% |
|
6 |
putative thymidylate synthase |
thyA |
EC:2.1.1.45 |
Catalyzes the reductive methylation of 2'-deoxyuridine-5'-monophosphate (dUMP) to 2'-deoxythymidine-5'-monophosphate (dTMP) |
thymidylate synthase, 1e-54, 35.51% |
|
7 |
putative thymidylate kinase |
tmk |
EC:2.7.4.9 |
Phosphorylation of dTMP to form dTDP in both de novo and salvage pathways of dTTP synthesis |
thymidylate kinase, 7e-08, 28.92% |
Structure Prediction of the Uridylate Kinase
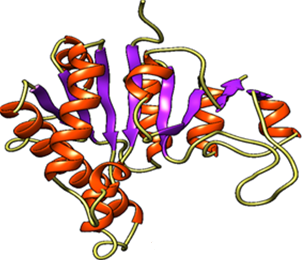
The 3D structure of uridylate kinase of Burkholderia pseudomallei strain K96243 is not available in the PDB. Hence, the alpha fold server was used to obtain the 3D structure and visualized it. The key binding sites were obtained from the UniProt server (https://www.uniprot.org/uniprotkb/Q63T14/entry) and are shown in Figure 2.
|
|
|
|
a) |
b) |
|
|
|
|
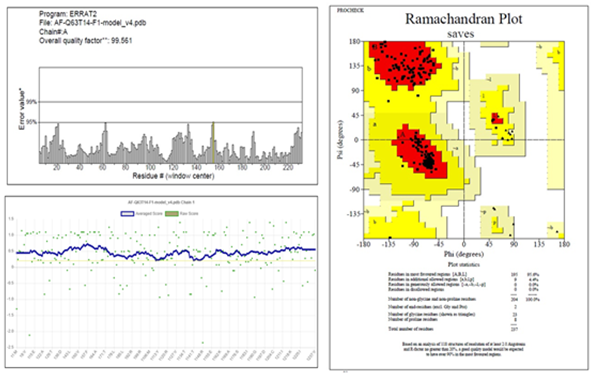
Figure 2. a) Predicted 3D structure of UMP kinase enzyme and b) binding residues ATP binding residues are 11-14, 54,58, 161,167,170 (Red color highlights), UMP binding sites are 53, 73, 134-141 (Blue color highlights). The lower section describes validation features of the UMP kinase 3D model. |
|
Table 2. Ligand details selected for docking studies
|
S. N |
Compound (CID) |
name |
DOCKING SCORE (Kcal/mole) |
|
1 |
25390206 |
N-benzyl-2-[(2S,3R,4S,5R)-3,4-dihydroxy-5-(methanesulfonamidomethyl)oxolan-2-yl]-N methylacetamide |
-7.3 |
|
2 |
28359984 |
2-(4-acetyl-3,5-dimethylpyrazol-1-yl)-N-[2-[(2-methoxyphenyl)methyl]pyrazol-3-yl]acetamide |
-7.3 |
|
3 |
284262 |
4-(2-Amino-4-(4-chlorophenyl)-7-oxo-5H-pyrrolo[3,4-d]pyrimidin-6(7H)-yl)butanoic acid |
-8.1 |
|
4 |
284264 |
4-[2-amino-4-(4-methoxyphenyl)-7-oxo-5H-pyrrolo[3,4-d]pyrimidin-6-yl]butanoic acid |
-7.8 |
|
5 |
6133 |
[[(2R,3S,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl] methoxy-hydroxyphosphoryl] phosphono hydrogen phosphate (UTP) |
-8.7 |
|
6 |
8900795 |
1-[2-[[5-oxo-4-[[(2R)-oxolan-2-yl]methyl]-1H-1,2,4-triazol-3-yl]sulfanyl]acetyl]piperidine-4-carboxylate |
-6.9 |
The 3D structure of the molecule was predicted, and the ATP and UMP binding residues were highlighted (Figure 2). The model was further subjected to validation by using different parameters. The Ramachandran plot of the model shows 96.5% of residues are in favorable regions. The analysis of the model by the Errat tool generated a quality factor of 99.561, and a satisfactory level of the plot was obtained by Verify 3D tool (Figure 2). The overall result indicates that the model quality is good and can be considered for further study. Molecular docking by AutodockVina tool predicted the UTP, the enzyme's natural inhibitor having the highest binding affinity (docking score = -8.7 Kcal/mole). However, another molecule, CID 284262, also showed a comparatively good docking score (-8.1 Kcal/mole) (Table 2).
|
|
|
a) |
|
|
|
b) |
|
|
|
c) |
|
|
|
d) |
|
|
|
e) |
|
|
|
f) |
|
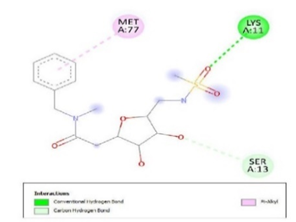
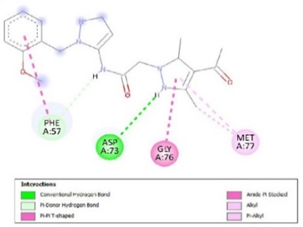
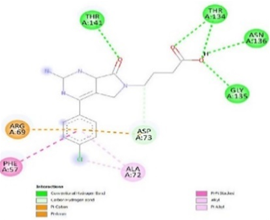
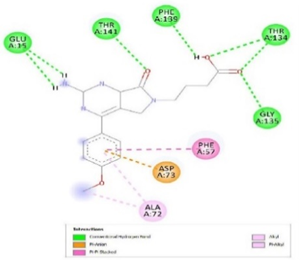
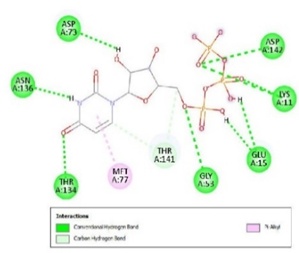
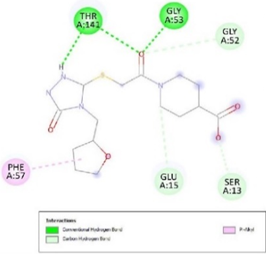
Figure 3. Ligand Binding site and interaction details |
|
|
|
a) |
|
|
|
b) |
|
|
|
c) |
|
|
|
d) |
|
|
|
e) |
|
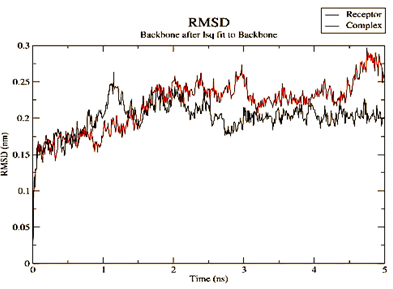
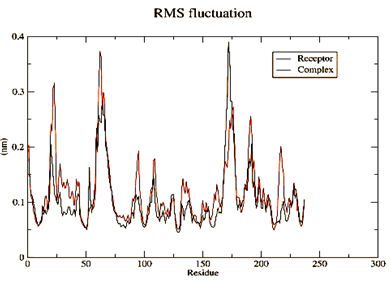
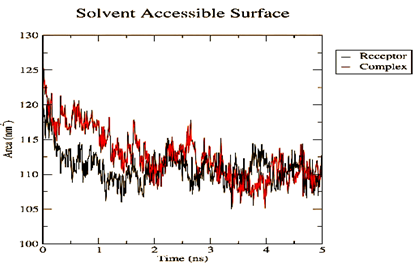
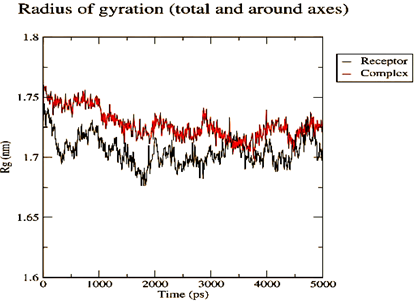
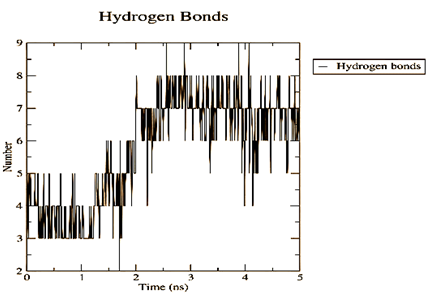
Figure 4. Parameters computed after MD simulation of UMPkinase -CID284262 complex (red color and black color represent simulation result of ligand-receptor complex and receptor only respectively) |
The molecule CID 284262 formed the four hydrogen bonds with the receptor as Thr 134, Gly135, Asn 136, and Thr 141 (Figure 3c), which covers the UMP binding site of the enzyme (Figure 3). During molecular dynamics simulation, the RMSD values of each frame corresponding to time are given in Figure 4. An initial sharp increase in the RMSD values indicated that, for a period of up to 2 ns, the RMSD values of the ligand-receptor complex rose gradually. Finally, at five ns, the values were observed to be decreased. The RMSD values were within 0.3 nm, demonstrating that the backbone of the protein molecule did not fluctuate much. The amino acid residue fluctuation was computed from the RMSF plot generated during the 5 ns MD simulation. The mean RMSF value indicated that each amino acid residues were stable of the protein during MD simulation. The Rg values indicate the rigidity of the protein system and were analyzed during the MD simulation. The data show that the Rg value was very close to the maximum, indicating the system's stability throughout the five ns MD simulation (Pingale & Amrutkar, 2021). Also, there is a decreasing trend in the SASA value was observed. Similarly, the hydrogen bonding pattern was observed to be strengthened during the MD simulation. Therefore, overall, the results confirm the stability of the UMP Kinase- CID 284262 complex molecular system considered for the present MD simulation study (Abdulazeez, 2019; Islam et al., 2021; Biharee et al., 2022).
Table 3. Toxicity features and some significant ADMET properties of the molecules
|
S. N |
Compound (CID) |
GI absorption |
BBB permeant |
Synthetic accessibility |
Solubility |
Lead- likeness |
Toxicity level |
|
1 |
25390206 |
High |
No |
4.08 |
Soluble |
No |
5 |
|
2 |
28359984 |
High |
No |
3.19 |
Moderately soluble |
No |
4 |
|
3 |
284262 |
High |
No |
2.63 |
Moderately soluble |
Yes |
4 |
|
4 |
284264 |
High |
No |
2.71 |
Moderately soluble |
Yes |
4 |
|
5 |
6133 |
Low |
No |
5.02 |
Soluble |
No |
6 |
|
6 |
8900795 |
Low |
No |
3.17 |
Soluble |
No |
4 |
The toxicity profiling of compounds showed the natural inhibitor UTP is completely non-toxic; one compound CID 25390206, was observed as under toxicity level 5, and other molecules are under Toxicity level 4 (harmful if swallowed (300 < LD50 ≤ 2000) (Table 3) (Mustarichie & Saptarini, 2023). In addition, molecule 284262 has a LogP value of 2.72; hence there is a minor issue in the molecule's solubility (Banerjee et al., 2018). Several important ADMET properties were also computed from the Swiss ADME server and presented in Table 3. It shows that the Gi absorption of the proposed molecule is high and can be easily absorbed from the gastrointestinal tract by the cellular transport mechanism. Also, the molecule is not permeable to the blood-brain barrier (BBB) shield of the brain. Least synthetic accessibility is also another advantage of the drug molecule. However, solubility was observed as an issue when considering it as a drug molecule (Daina et al., 2017; Bergström & Larsson, 2018).
Fassy et al., in their work, selected the UMP kinase enzyme of Streptococcus pneumoniae as an effective drug target for the antibacterial compound development. The work also explained the essential function of the UMP kinase in the catalyzation of UMP by ATP phosphorylation to produce UDP and ADP (Fassy et al., 2004). Chong et al. compared the genome of Burkholderia pseudomallei with different species and identified 312 essential genes (probable drug targets) of the pathogen using computational methods (Chong et al., 2006). Egeblad‐Welin et al. derived the crystallographic structure of the UMP kinase from the pathogen Ureaplasma parvum, which show similarity in the 3D structural fold with other bacterial and archaeal UMP kinase enzyme. The research provides insight into enzyme selection as a potential target, and the possibility of developing inhibitor compounds has been discussed (Egeblad‐Welin et al., 2007). Rostirolla et al. reviewed that the UMP kinase enzyme can be suitably used as the drug target in the case of Mycobacterium tuberculosis, based on which design of novel inhibitor molecules are possible (Rostirolla et al., 2009). In another work, Rostirolla et al. described that the uridine 5′-monophosphate kinase (encoded by the pyrH gene) is involved in de novo and salvage synthesis of nucleotide biosynthesis in case of Mycobacterium tuberculosis bacteria. Since the enzyme does not show homology with the eukaryotic (human) counterparts, specific inhibitors as anti-tubercular agents could be designed (Rostirolla et al., 2011). Yoshida et al., in their research, showed that the compound PYRH-1 is the effective inhibitor of the UMPkinase enzyme in the case of both Streptococcus pneumoniae and Haemophilus influenza bacteria (Yoshida et al., 2012). Moule et al. conducted a genome-wide study to screen the drug targets in the B. pseudomallei genome. In their research, they identified 505 essential genes in B. pseudomallei K96243. Three genes are predicted to be crucial, pyrH (encodes uridylate kinase), accA (encodes acetyl-CoA carboxylase carboxyltransferase subunit alpha), and sodB (encodes superoxide dismutase) (Moule et al., 2014). Ross et al. screened a compound library containing 400 inhibitor molecules and predicted seven potential compounds that show anti-bacterial activity against B. pseudomallei K96243 (Ross et al., 2018). Khan et al. reported about 45 essential, unique, human non-homologous proteins of Burkholderia pseudomallei K96243 using a subtractive-genomics approach. Further screening resulted in 36 proteins selected for molecular docking with 14 antibiotics followed by 100ns MD simulation. Finally, two enzymes, dihydroneopterin aldolase, and phosphoribosyl transferase were predicted as novel drug targets. Also, the research identified the antibiotics cefiderocol and tetracycline show the best affinity towards the selected target (Khan et al., 2022). Recently, Walter et al. studied the molecular mechanism and effect of UTP binding to the UMP kinase enzyme of Mycobacterium tuberculosis bacteria by using X-ray crystallography and cryo-electron microscopy. They reported that the UTP binding causes substantial flexibility in the Mg-ATP-binding domain of the enzyme. The fact can be helpful and might be considered while targeting the bacterial UMP kinase enzyme in drug discovery (Walter et al., 2022).
Conclusion
Burkholderia pseudomallei infection is considered a global threat due to its ability to cause melioidosis. This work used a bioinformatics pipeline to study the potential targets and inhibitors of Burkholderia pseudomallei strain K96243. First, the computational method was used to analyze the pyrimidine biosynthetic pathway, and the UMP kinase enzyme was selected as the suitable drug target for the bacteria. After prediction and validation of the 3D structure followed by molecular docking, inhibitor compounds UTP (CID 6133) and (CID 284262) was obtained as potential inhibitor molecule. Furthermore, the stability of the binding complex (UMP kinase - CID 284262) was analyzed by 5 ns MD simulation and accessing parameters like RMSD, RMSF, Rg, SASA value, and hydrogen bonding pattern. Finally, toxicity features were computed for the selected ligand molecules with some important features. The analysis predicted that the compound PubChem CID 284262 is effective and can be used to disrupt the function of the UMP Kinase enzyme. Further, the rational design in the molecule might be helpful to reduce the toxicity and solubility issues. Later on, synthesis and pre-clinical studies of the designed compound with Burkholderia pseudomallei strain K96243 are recommended for the therapeutic management of melioidosis disease. However, this is a computational prediction, hence, experimental verification is necessary to validate the fact.
Acknowledgments: None
Conflict of interest: None
Financial support: None
Ethics statement: None
Abdulazeez, S. (2019). Molecular simulation studies on B-cell lymphoma/leukaemia 11A (BCL11A). American Journal of Translational Research, 11(6), 3689-3697.
Aleidi, S. A., Alosaimi, N. S., Aljumah, S. M., Alabdulmunim, R. A., & Alhussain, B. (2022). Assessment of Ionoseal's performance as a lining and sealing material in dental restorations: a comprehensive review. International Journal of Dental Research and Allied Sciences, 2(1), 13-19. doi:10.51847/iVCXX97n31
Arvind, A., Jain, V., Saravanan, P., & Mohan, C. G. (2013). Uridine monophosphate kinase as potential target for tuberculosis: From target to lead identification. Interdisciplinary Sciences, Computational Life Sciences, 5(4), 296-311. doi:10.1007/s12539-013-0180-y
Banerjee, P., Eckert, A. O., Schrey, A. K., & Preissner, R. (2018). ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Research, 46(W1), W257-W263. doi:10.1093/nar/gky318
Beji, F., Al Harbi, R., & Naqvi, A. (2020). In-silico Molecular Docking, ADME and Drug Likeness Predictions of Some α-alkylidene-β-ethoxycarbonyl Cyclopentanones. International Journal of Pharmaceutical and Phytopharmacological Research, 10(4), 126-132.
Bergström, C. A. S., & Larsson, P. (2018). Computational prediction of drug solubility in water-based systems: Qualitative and quantitative approaches used in the current drug discovery and development setting. International Journal of Pharmaceutics, 540(1–2), 185-193. doi:10.1016/j.ijpharm.2018.01.044
Biharee, A., Yadav, A., Jangid, K., Singh, Y., Kulkarni, S., Sawant, D. M., Kumar, P., Thareja, S., & Jain, A. K. (2022). Flavonoids as promising anticancer agents: An in silico investigation of ADMET, binding affinity by molecular docking and molecular dynamics simulations. Journal of Biomolecular Structure and Dynamics, 1-12. doi:10.1080/07391102.2022.2126397
Chong, C. E., Lim, B. S., Nathan, S., & Mohamed, R. (2006). In silico analysis of Burkholderia pseudomallei genome sequence for potential drug targets. In Silico Biology, 6(4), 341-346.
Currie, B. J. (2022). Melioidosis and Burkholderia pseudomallei: Progress in epidemiology, diagnosis, treatment and vaccination. Current Opinion in Infectious Diseases, 35(6), 517-523. doi:10.1097/QCO.0000000000000869
Daina, A., Michielin, O., & Zoete, V. (2017). SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports, 7(1), 42717. doi:10.1038/srep42717
Daivasigamani, S., Chidambaranathan, A. S., & Balasubramanium, M. (2022). A systematic review on the color stability of maxillofacial silicone materials after disinfection and aging procedures. International Journal of Dental Research and Allied Sciences, 2(1), 8-12. doi:10.51847/8qZssQqjrK
Di Fiore, A., De Luca, V., Langella, E., Nocentini, A., Buonanno, M., Monti, S. M., Supuran, C. T., Capasso, C., & De Simone, G. (2022). Biochemical, structural, and computational studies of a γ-carbonic anhydrase from the pathogenic bacterium Burkholderia pseudomallei. Computational and Structural Biotechnology Journal, 20, 4185-4194. doi:10.1016/j.csbj.2022.07.033
Egeblad‐Welin, L., Welin, M., Wang, L., & Eriksson, S. (2007). Structural and functional investigations of Ureaplasma parvum UMP kinase–A potential antibacterial drug target. FEBS Journal, 274(24), 6403-6414. doi:10.1111/j.1742-4658.2007.06157.x
Ekakoro, J. E., Lubega, A., Kayaga, E. B., Ndoboli, D., Bluhm, A. P., Wampande, E. M., Blackburn, J. K., Havas, K. A., & Norris, M. H. (2022). An investigation of Burkholderia pseudomallei seroprevalence in market pigs slaughtered at selected pig abattoirs in Uganda. Pathogens, 11(11), 1363. doi:10.3390/pathogens11111363
Fassy, F., Krebs, O., Lowinski, M., Ferrari, P., Winter, J., Collard-Dutilleul, V., & Salahbey Hocini, K. (2004). UMP kinase from Streptococcus pneumoniae: Evidence for co-operative ATP binding and allosteric regulation. Biochemical Journal, 384(3), 619-627. doi:10.1042/BJ20040440
Gassiep, I., Armstrong, M., & Norton, R. (2020). Human melioidosis. Clinical Microbiology Reviews, 33(2), e00006-19. doi:10.1128/CMR.00006-19
Ghahremanian, S., Rashidi, M. M., Raeisi, K., & Toghraie, D. (2022). Molecular dynamics simulation approach for discovering potential inhibitors against SARS-CoV-2: A structural review. Journal of Molecular Liquids, 354, 118901. doi:10.1016/j.molliq.2022.118901
Inglis, T. J. J., Mee, B. J., & Chang, B. J. (2001). The environmental microbiology of melioidosis. Reviews in Medical Microbiology, 12(1), 13-20. doi:10.1097/00013542-200101000-00002
Islam, R., Parves, M. R., Paul, A. S., Uddin, N., Rahman, M. S., Mamun, A. A., Hossain, M. N., Ali, M. A., & Halim, M. A. (2021). A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 39(9), 3213-3224. doi:10.1080/07391102.2020.1761883
Khan, M. A. S., Miah, M. I., & Rahman, S. R. (2022). Subtractive proteomic analysis for identification of potential drug targets and vaccine candidates against Burkholderia pseudomallei K96243. Informatics in Medicine Unlocked, 35, 101127. doi:10.1016/j.imu.2022.101127
Khattab, M. A., Abou Kamar, M., Darweesh, A., & Deshmukh, A. (2022). Melioidosis an imported infection in Qatar: Case series and literature review. Advances in Infectious Diseases, 12(2), 175-192.
Kumari, S., & Tripathi, P. (2021). Nucleotide metabolism pathway: The Achilles’ heel for bacterial pathogens. Current Science, 120(9), 1458-1463. doi:10.18520/cs/v120/i9/1458-1463
Leung, C. C. D., Ngai, C. M., Wong, C. K., & Chan, Y. H. (2023). A rare case of melioidosis presenting as pericarditis and pneumonia in a patient with poorly controlled diabetes mellitus. Respirology Case Reports, 11(4), e01119. doi:10.1002/rcr2.1119
Morici, L., Torres, A. G., & Titball, R. W. (2019). Novel multi-component vaccine approaches for Burkholderia pseudomallei. Clinical and Experimental Immunology, 196(2), 178-188. doi:10.1111/cei.13286
Moule, M. G., Hemsley, C. M., Seet, Q., Guerra-Assunção, J. A., Lim, J., Sarkar-Tyson, M., & Wren, B. W. (2014). Genome-wide saturation mutagenesis of Burkholderia pseudomallei K96243 predicts essential genes and novel targets for antimicrobial development. mBio, 5(1), e00926-13.
Mustarichie, R., & Saptarini, N. M. (2023). Sirih (Piper betle) folium as new candidate for anti-herpes virus: In-silico study. Journal of Advanced Pharmacy Education and Research, 13(1), 46-50. doi:10.51847/mcuhssaHlU
Perveen, S., & Sharma, R. (2022). Screening approaches and therapeutic targets: The two driving wheels of tuberculosis drug discovery. Biochemical Pharmacology, 197, 114906. doi:10.1016/j.bcp.2021.114906
Pingale, P. L., & Amrutkar, S. V. (2021). Quercetin loaded rifampicin-floating microspheres for improved stability and in-vitro drug release. Pharmacophore. 12(3), 95-99. doi:10.51847/yBXnl2bSUH
Ross, B. N., Myers, J. N., Muruato, L. A., Tapia, D., & Torres, A. G. (2018). Evaluating new compounds to treat Burkholderia pseudomallei infections. Frontiers in Cellular and Infection Microbiology, 8, 210. doi:10.3389/fcimb.2018.00210
Rostirolla, D. C., Breda, A., Basso, L. A., & Santos, D. S. (2009). The protein UMP kinase from Mycobacterium tuberculosis [Mostra de pesquisa da Pós-Graduação – PUCRS], IV, 107–109 (European Commission 2.7, 4.-) as target for development of new drugs.
Rostirolla, D. C., Breda, A., Rosado, L. A., Palma, M. S., Basso, L. A., & Santos, D. S. (2011). UMP kinase from Mycobacterium tuberculosis: Mode of action and allosteric interactions, and their likely role in pyrimidine metabolism regulation. Archives of Biochemistry and Biophysics, 505(2), 202-212. doi:10.1016/j.abb.2010.10.019
Satpathy, R. (2020). Application of molecular docking methods on endocrine disrupting chemicals: A review. Journal of Applied Biotechnology Reports, 7(2), 74-80.
Savelkoel, J., Dance, D. A. B., Currie, B. J., Limmathurotsakul, D., & Wiersinga, W. J. (2022). A call to action: Time to recognise melioidosis as a neglected tropical disease. Lancet. Infectious Diseases, 22(6), e176-e182. doi:10.1016/S1473-3099(21)00394-7
Somprasong, N., Yi, J., Hall, C. M., Webb, J. R., Sahl, J. W., Wagner, D. M., Keim, P., Currie, B. J., & Schweizer, H. P. (2021). Conservation of resistance-nodulation-cell division efflux pump-mediated antibiotic resistance in Burkholderia cepacia complex and Burkholderia pseudomallei complex species. Antimicrobial Agents and Chemotherapy, 65(9), e0092021. doi:10.1128/AAC.00920-21
Sridharan, S., Princess, I. B., & Ramakrishnan, N. (2021). Melioidosis in critical care: A review. Indian Journal of Critical Care Medicine, 25(Suppl. 2), S161-S165. doi:10.5005/jp-journals-10071-23837
Uddin, R., Siraj, B., Rashid, M., Khan, A., Ahsan Halim, S., & Al-Harrasi, A. (2020) Genome subtraction and comparison for the identification of novel drug targets against mycobacterium avium subsp. hominissuis. Pathogens, 9(5), 368. doi:10.3390/pathogens9050368
Walter, P., Mechaly, A., Bous, J., Haouz, A., England, P., Lai‐Kee‐Him, J., Ancelin, A., Hoos, S., Baron, B., Trapani, S., et al. (2022). Structural basis for the allosteric inhibition of UMP kinase from Gram-positive bacteria, a promising antibacterial target. FEBS Journal, 289(16), 4869-4887. doi:10.1111/febs.16393
Wiersinga, W. J., Virk, H. S., Torres, A. G., Currie, B. J., Peacock, S. J., Dance, D. A. B., & Limmathurotsakul, D. (2018). Melioidosis. Nature Reviews. Disease Primers, 4(1), 17107. doi:10.1038/nrdp.2017.107
Wyllie, J. A., McKay, M. V., Barrow, A. S., & Soares da Costa, T. P. (2022). Biosynthesis of uridine diphosphate N‐Acetylglucosamine: An underexploited pathway in the search for novel antibiotics? IUBMB Life, 74(12), 1232-1252. doi:10.1002/iub.2664
Yoshida, T., Nasu, H., Namba, E., Ubukata, O., & Yamashita, M. (2012). Discovery of a compound which acts as a bacterial UMP kinase PyrH inhibitor. FEMS Microbiology Letters, 330(2), 121-126. doi:10.1111/j.1574-6968.2012.02546.x
Zueter, A. M., Sawan, H. M., Zaiter, A., & Harun, A. (2022). Current protocols in laboratory diagnosis, genotyping, and treatment of Burkholderia pseudomallei. Clinical Microbiology Newsletter, 44(3), 23-31. doi:10.1016/j.clinmicnews.2022.01.003